Пралуэнт — инструкция по применению
Синонимы, аналоги
Статьи
Описание предварительно заполненных одноразовых шприц-ручек и инструкция по их использованию
Описание предварительно заполненных одноразовых шприцев и инструкция по их использованию
Регистрационный номер:
ЛП-004078 — 240317
Торговое наименование препарата:
Пралуэнт
Международное непатентованное наименование:
алирокумаб
Лекарственная форма:
раствор для подкожного введения.
Состав:
В одном шприце/шприц-ручке с дозировкой 75 мг/мл содержится:
активное вещество: алирокумаб — 75,0 мг;
вспомогательные вещества: L-гистидин и L-гистидина гидрохлорида моногидрат — 1,241* мг, сахароза — 100,0 мг, полисорбат-20 — 0,1 мг, вода для инъекций — до 1,0 мл.
В одном шприце/шприц-ручке с дозировкой 150 мг/мл содержится:
активное вещество: алирокумаб — 150,0 мг;
вспомогательные вещества: L-гистидин и L-гистидина гидрохлорида моногидрат — 0,931* мг, сахароза — 100,0 мг, полисорбат-20 — 0,1 мг, вода для инъекций — до 1,0 мл.
* — суммарное количество L-гистидина и L-гистидина гидрохлорида моногидрата в пересчете на L-гистидин.
Описание:
Прозрачная или слегка опалесцирующая, бесцветная или желтоватого цвета жидкость.
Фармакотерапевтическая группа:
полностью гуманизированное моноклональное антитело (IgGl). Ингибитор пропротеиновой конвертазы субтилизинкексин типа 9 (PCSK9)
Код АТХ:
С10АХ14
Фармакологические свойства
Фармакодинамика
Алирокумаб является полностью гуманизированным моноклональным согласовано антителом [изотип иммуноглобулинов G1 (IgGl)], мишенью которого является фермент пропротеиновая конвертаза субтилизин-кексин типа 9 (PCSK9). Алирокумаб производится с помощью технологии рекомбинантной ДНК с использованием суспензионной культуры клеток яичника китайского хомячка.
Алирокумаб имеет молекулярную массу приблизительно 146 кДа.
Механизм действия
PCSK9 связывается с рецепторами липопротеинов низкой плотности (Р-ЛПНП) на поверхности гепатоцитов, способствуя деградации Р-ЛПНП в печени. Р-ЛПНП являются главными рецепторами, которые выводят из системного кровотока циркулирующие ЛПНП, поэтому уменьшение количества Р-ЛПНП при связывании их с PCSK9 приводит к повышению концентрации холестерина ЛПНП (ХС-ЛПНП) в крови. Ингибируя связывание PCSK9 с Р-ЛПНП, алирокумаб увеличивает количество Р-ЛПНП для выведения ЛПНП, снижая, таким образом, концентрации ХС-ЛПНП в крови.
Р-ЛПНП также связывают богатые триглицеридами (ТГ) ремнантные липопротеины очень низкой плотности (ЛПОНП) и липопротеины промежуточной плотности (ЛППП). Поэтому лечение алирокумабом может снижать концентрации этих ремнантных липопротеинов, о чем свидетельствует их уменьшение в аполипопротеине В (Апо В), холестерине липопротеинов, не являющихся липопротеинами высокой плотности (ХС-ЛПнеВП) и ТГ. Алирокумаб также снижает концентрации липопротеинов а (Лп(а)), являющихся формой ЛПНП, которые связаны с аполипопротеином (а). Однако было показано, что Р-ЛПНП имеют низкую аффинность к Лп(а), в связи с чем точный механизм, с помощью которого алирокумаб снижает Лп(а), полностью не установлен.
Генетические исследования
В генетических исследованиях, проведенных у человека, были выявлены разновидности гена PCSK9 с мутациями потери или повышения функции. У пациентов с одним аллелем PCSK9 с мутацией потери функции отмечались более низкие концентрации ХС-ЛПНП, которые коррелировали со значительно более низкой частотой развития ишемической болезни сердца. У некоторых пациентов были выявлены мутации потери функции в двух аллелях, и у них отмечались очень низкие концентрации ХС-ЛПНП в крови с концентрациями в крови ХС-ЛПВП и ТГ в нормальном диапазоне. Наоборот, мутации повышения функции в гене PCSK9 были выявлены у пациентов с повышенными концентрациями ХС-ЛПНП в крови и клиническим диагнозом семейной гиперхолестеринемии.
Наблюдательный анализ показал, что без лечения концентрации ХС-ЛПНП в крови у пациентов с мутациями повышения функции в гене PCSK9 находились в диапазоне, подобном наблюдавшемуся у пациентов с более часто встречающимися мутациями, вызывающими гетерозиготную форму семейной гиперхолестеринемии (такими как мутации в гене Р-ЛПНП), демонстрируя центральную роль фермента PCSK9 в метаболизме ХС-ЛПНП и его концентрациях в крови. В многоцентровом двойном слепом, плацебо-контролируемом исследовании продолжительностью 14 недель 13 пациентов с гетерозиготной формой семейной гиперхолестеринемией, связанной с мутацией повышения функции в гене PCSK9, были рандомизированы в 2 группы: группу, получающую алирокумаб в дозе 150 мг 1 раз в 2 недели, и группу, получающую плацебо. Среднее значение концентрации ХС-ЛПНП в крови составляло 151,5 мг/дл. На второй неделе лечения среднее значение снижения исходной концентрации ХС-ЛПНП в крови составило 62,5 % в группе пациентов, получавших алирокумаб, по сравнению с 8,8 % у пациентов, получавших плацебо. На 8-й неделе лечения среднее значение снижения концентрации ХС-ЛПНП в крови от исходного значения у всех пациентов, получавших алирокумаб, составило 72,4 %.
Фармакодинамические свойства
Алирокумаб является полностью гуманизированным моноклональным антителом, которое подавляет активность PCSK9 как в исследованиях in vitro, так и в системах моделей in vivo. Большое количество исследований, проведенных у человека и животных, продемонстрировали центральную роль, которую играют повышенные концентрации ХС-ЛПНП в крови в начале и прогрессировании атеросклероза. Другие липопротеины, содержащие Апо В-100, особенно богатые триглицеридами ремнантные липопротеины (образовавшиеся из ЛПОНП и ЛППП) и Лп(а), также считаются способствующими развитию атеросклероза. Однако проведенные до настоящего времени исследования не выявили независимого влияния снижения концентраций этих липопротеинов на сердечно-сосудистую заболеваемость и смертность.
В исследованиях in vitro, алирокумаб не индуцировал антитело-зависимую клеточно-опосредованную токсичность и комплемент-зависимую цитотоксичность (Fc-опосредованную эффекторную функцию), как в присутствии, так и в отсутствии PCSK9. У алирокумаба, связанного с PCSK9, не наблюдалось образования нерастворимых иммунных комплексов, способных связывать протеины комплемента.
Клиническая эффективность/клинические исследования
Эффективность алирокумаба была изучена в 10 исследованиях III фазы (5 плацебо-контролируемых и 5 эзетимиб-контролируемых исследований), включавших 5296 рандомизированных пациентов с гиперхолестеринемией (несемейной и гетерозиготной формой семейной) или смешанной гиперхолестеринемией, из них 3188 пациентов были рандомизированы для приема алирокумаба. Три из этих 10 исследований были проведены исключительно у пациентов с гетерозиготной формой семейной гиперхолестеринемии. Большинство пациентов в программе клинических исследований III фазы принимали одновременно липид-модифицирующую терапию, состоящую из максимально переносимых доз статинов в сочетании или без сочетания с другими липид-модифицирующими видами лечения и 1 г, имели высокий и очень высокий сердечно-сосудистыи риск.
Два исследования были проведены у пациентов, которые не получали одновременно статины, включая одно исследование у пациентов с документированной непереносимостью статинов.
Два исследования (LONG TERM и HIGH FH), включающие в общей сложности 2416 пациентов, были проведены только с дозой 150 мг 1 раз в 2 недели. Восемь исследований были проведены с дозой 75 мг 1 раз в 2 недели и критерием для повышения дозы до 150 мг 1 раз в 2 недели на 12-й неделе было недостижение пациентами целевого значения концентрации ХС-ЛПНП в крови, основанного на степени их сердечно-сосудистого риска на 8-й неделе лечения.
Исходные демографические характеристики были хорошо сбалансированными между группами алирокумаба и контроля. Возраст пациентов во всех исследованиях находился в диапазоне от 18 до 89 лет (средний возраст составлял 60 лет); 38 % пациентов были женского пола, большинство пациентов были белой расы, 5 % были негроидной расы, 2 % были представителями азиатской расы; среднее значение индекса массы тела (ИМТ) составляло 30 кг/м2. В клинических исследованиях III фазы 31 % пациентов имели сахарный диабет 2 типа и 64 % пациентов имели в анамнезе ишемическую болезнь сердца.
Главной конечной точкой эффективности во всех клинических исследованиях III фазы было среднее значение снижения концентрации ХС-ЛПНП в крови на 24-й неделе по сравнению с плацебо и эзетимибом. Все исследования соответствовали их главной конечной точке.
В целом, применение алирокумаба приводило также к статистически значимо большему значению процента снижения концентраций ОХ, ХС-ЛПнеВП, Апо В и Лп(а) по сравнению с плацебо/эзетимибом, независимо от того получали или нет пациенты статины. Алирокумаб также снижал концентрации ТГ и увеличивал концентрации ХС-ЛПВП и Апо А-1 по сравнению с плацебо.
Снижение концентрации ХС-ЛПНП в крови наблюдалось во всех возрастных группах, у лиц обоего пола, при разных показателях индекса массы тела (ИМТ), у лиц разных рас, и с разными исходными концентрациями ХС-ЛПНП в крови. Результаты по эффективности были единообразными у пациентов с гетерозиготной формой семейной гиперхолестеринемии, без гетерозиготной семейной гиперхолестеринемии, у пациентов со смешанной дислипидемией и пациентов с сахарным диабетом. Снижение концентраций ХС-ЛПНП в крови было единообразным, независимо от того, принимались или не принимались пациентами одновременно статины, и от доз последних.
В группе алирокумаба по сравнению с группами плацебо или эзетимиба на 12-й и 24-й неделе статистически значимо у более высокого процента пациентов была достигнута концентрация ХС-ЛПНП в крови, составляющая <70 мг/дл.
В исследованиях с использованием схем титрования дозы, основанных на соответствующих критериях, большинство пациентов достигали целевого значения концентрации ХС-ЛПНП в крови (основанного на их степени сердечно-сосудистого риска) на дозе 75 мг 1 раз в 2 недели, и большинство пациентов продолжали лечение в дозе алирокумаба 75 мг 1 раз в 2 недели. Липидоснижающий эффект алирокумаба наблюдался в пределах 15 дней после применения первой дозы, достижение максимального эффекта наблюдалось приблизительно на 4 неделе. Эффективность сохранялась на протяжении всей продолжительности исследуемого лечения (до 78 недель в исследовании LONG TERM).
После прекращения введения алирокумаба не наблюдалось «синдрома отмены» и концентрация ХС-ЛПНП в крови постепенно возвращалась к исходным показателям.
Фармакокинетика
Абсорбция
После подкожного введения препарата Пралуэнт в дозе опт 50 мг до 300 мг среднее время достижения максимальной концентрации алирокумаба в сыворотке крови составляло 3-7 дней.
Фармакокинетика алирокумаба после однократного подкожного введения в дозе 75 мг в область живота, бедра или плеча была подобной. По данным популяционного анализа фармакокинетических показателей абсолютная биодоступность алирокумаба после подкожного введения составляла 85 %.
Незначительно большее (2,1-2,7-кратное), чем пропорциональное дозе, увеличение концентраций алирокумаба, наблюдалось при двукратном увеличении дозы с 75 мг до 150 мг 1 раз в 2 недели.
Равновесное состояние достигалось после введения 2-3 доз с двукратным коэффициентом накопления.
Распределение
После внутривенного введения объем распределения алирокумаба составлял 0,04-0,05 л/кг, что указывает на распределение алирокумаба, главным образом, в системе кровообращения.
Метаболизм
Специальные исследования метаболизма не проводились, так как алирокумаб является белком. Предполагается, что алирокумаб расщепляется на небольшие пептиды и отдельные аминокислоты.
Выведение
У алирокумаба наблюдались 2 фазы выведения. В низких концентрациях элиминация происходит преимущественно через насыщаемую связь с мишенью (PCSK9), в то время как при более высоких концентрациях элиминация алирокумаба происходит преимущественно через ненасыщаемый протеолитический путь. По данным популяционного анализа фармакокинетических показателей средний период полувыведения алирокумаба составлял 17-20 дней у пациентов, получавших алирокумаб в монотерапии подкожно в дозах 75 мг 1 раз в 2 недели или 150 мг 1 раз в 2 недели. При одновременном применении со статинами, средний период полувыведения алирокумаба составлял 12 дней.
Особые группы пациентов
Пол
По данным популяционного анализа фармакокинетических показателей половая принадлежность пациентов не влияла на фармакокинетику алирокумаба.
Пациенты пожилого возраста
По данным популяционного анализа фармакокинетических показателей, возраст ассоциировался с небольшим различием в системной экспозиции алирокумаба в равновесном состоянии без влияния на его эффективность и безопасность.
Масса тела
По данным популяционного анализа фармакокинетических показателей масса тела оказывала незначительное влияние на системную экспозицию алирокумаба без влияния на его эффективность и безопасность.
Пациенты детского возраста
Фармакокинетические эффекты алирокумаба у пациентов детского возраста к настоящему моменту не изучались.
Нарушения функции печени
В клиническом исследовании I фазы при однократном подкожном введении алирокумаба в дозе 75 мг фармакокинетические показатели алирокумаба у пациентов с печеночной недостаточностью легкой и средней степени тяжести были аналогичными таковым у пациентов с нормальной функцией печени. Доступные данные по фармакокинетике алирокумаба у пациентов с печеночной недостаточностью тяжелой степени отсутствуют.
Нарушения функции почек
Так как нет сведений о том, что моноклональные антитела выводятся почками, не ожидается, что функциональное состояние почек повлияет на фармакокинетику алирокумаба. Популяционный анализ фармакокинетических показателей продемонстрировал, что нарушение функции почек легкой и средней степени тяжести не оказывало значимого воздействия на фармакокинетику алирокумаба. Данные по фармакокинетике алирокумаба у пациентов с тяжелыми нарушениями функции почек ограничены.
Расовая принадлежность
По данным популяционного анализа фармакокинетических показателей расовая принадлежность не оказывала влияния на фармакокинетику алирокумаба. После однократного подкожного введения алирокумаба в дозах 100-300 мг не наблюдалось значимых различий в системной экспозиции у здоровых добровольцев, являющихся японцами и представителями белой расы.
Показания к применению
Препарат Пралуэнт показан для длительного лечения взрослых пациентов с первичной гиперхолестеринемией (несемейной и гетерозиготной формой семейной гиперхолестеринемии) или смешанной дислипидемией, включая пациентов с сахарным диабетом 2 типа, в дополнение к диете, для снижения концентрации холестерина липопротеинов низкой плотности (ХС-ЛПНП), общего холестерина (общего-ХС), холестерина липопротеинов, не являющихся липопротеинами высокой плотности (ХС-ЛПнеВП), аполипопротеина В (Апо В), триглицеридов (ТГ) и липопротеина а (ЛПа) и повышения концентраций холестерина липопротеинов высокой плотности (ХС-ЛПВП) и аполипопротеина А-1 (Апо А-1). Препарат Пралуэнт показан:
- в комбинации со статинами (ингибиторами ГМГ-КоА-редуктазы) в сочетании или без сочетания с другой липид-модифицирующей терапией при невозможности достижения у пациентов целевой концентрации ХС-ЛПНП при приеме максимально допустимой дозы статинов;
- в монотерапии или как дополнение к другой, не содержащей статинов липид-модифицирующей терапии, у пациентов с непереносимостью статинов или при наличии противопоказаний к их применению.
Влияние препарата Пралуэнт на сердечно-сосудистую заболеваемость и смертность в настоящее время не установлено.
Противопоказания
- Повышенная чувствительность к алирокумабу или какому-либо вспомогательному веществу препарата.
- Беременность (эффективность и безопасность не установлены).
- Период грудного вскармливания (эффективность и безопасность не установлены).
- Детский возраст до 18 лет (эффективность и безопасность не установлены).
С осторожностью
- Почечная недостаточность тяжелой степени.
- Печеночная недостаточность тяжелой степени.
Применение при беременности и в период грудного вскармливания
Беременность
Отсутствуют данные по применению препарата Пралуэнт у беременных женщин.
Ожидается, что алирокумаб, как и другие антитела класса IgG, проникает через плацентарный барьер.
Во время беременности применение препарата Пралуэнт не рекомендуется.
Период грудного вскармливания
Неизвестно, проникает ли алирокумаб в грудное молоко у человека.
В связи с тем, что многие лекарственные препараты, и в том числе иммуноглобулины, проникают в грудное молоко у человека, применение препарата Пралуэнт у женщин в период грудного вскармливания не рекомендуется. При необходимости применения препарата Пралуэнт в данный период следует прекратить грудное вскармливание.
Способ применения и дозы
Начальная доза препарата Пралуэнт составляет 75 мг, которую вводят подкожно 1 раз в 2 недели. У пациентов, которым требуется большее снижение концентрации ХС-ЛПНП (> 60 %), начальная доза препарата Пралуэнт может составлять 150 мг, которую также вводят подкожно 1 раз в 2 недели.
Дозу препарата Пралуэнт следует подбирать индивидуально на основании таких параметров как исходные значения ХС-ЛПНП, цели терапии и ответ пациента на лечение. Концентрации липидов в крови можно оценивать через 4 недели после начала лечения или титрования дозы и проводить соответствующую коррекцию дозы.
В случае пропуска дозы пациент должен получить инъекцию, как можно скорее, и затем продолжить лечение через 2 недели со дня пропущенной дозы.
Особые группы пациентов
Дети
Безопасность и эффективность применения препарата Пралуэнт у детей в возрасте до 18 лет не установлены.
Пациенты пожилого возраста
У пациентов пожилого возраста коррекции дозы препарата Пралуэнт не требуется (см. раздел «Особые указания)».
Печеночная недостаточность
У пациентов с печеночной недостаточностью легкой или средней степени тяжести коррекции дозы препарата Пралуэнт не требуется. Данные по применению препарата Пралуэнт у пациентов с печеночной недостаточностью тяжелой степени отсутствуют.
Почечная недостаточность
У пациентов с почечной недостаточностью легкой или средней степени тяжести коррекции дозы препарата Пралуэнт не требуется. Данные по применению препарата у пациентов с почечной недостаточностью тяжелой степени ограничены.
Масса тела
Не требуется коррекции режима дозирования в зависимости от массы тела пациентов.
Правила введения препарата
Препарат Пралуэнт применяют в виде подкожных инъекций, проводимых в области бедра, живота или плеча, с помощью одноразовой предварительно заполненной шприц-ручки или одноразового предварительно заполненного шприца.
Рекомендуется менять места инъекций при проведении каждой инъекции.
Препарат Пралуэнт не следует вводить в области активных кожных заболеваний или повреждений, таких как солнечные ожоги, кожная сыпь, воспаления кожи или кожные инфекции.
Не следует вводить препарат Пралуэнт в то же место, в которое вводились другие лекарственные препараты.
Побочное действие
Представленные ниже данные по безопасности отражают применение алирокумаба у 3340 пациентов, большинство из которых имели высокий или очень высокий риск развития сердечно-сосудистых заболеваний, и которые получали алирокумаб в дозе 75 мг или 150 мг в виде подкожных инъекций 1 раз в 2 недели с продолжительностью лечения до 18 месяцев (включая 2408 пациентов, получавших лечение алирокумабом в течение 52 недель, и 639 пациентов, получавших лечение алирокумабом в течение не менее 76 недель). Данные по безопасности основаны на объединенных результатах 9 плацебо-контролируемых исследований (4 исследования фазы II и 5 исследований фазы III (все исследования у пациентов, одновременно принимающих статины), и 5 контролируемых эзетимибом исследований фазы III (в 3-х из которых пациенты одновременно принимали статины).
Наиболее частыми нежелательными реакциями (>1 % пациентов, получавших препарат Пралуэнт) были реакции в месте введения препарата, субъективные симптомы и объективные признаки со стороны верхних дыхательных путей и кожный зуд.
Наиболее частыми нежелательными реакциями, приводящими к прекращению лечения у пациентов, получавших препарат Пралуэнт, были реакции в месте введения препарата.
Не наблюдалось различий в отношении профиля безопасности между двумя дозами (75 мг 1 раз в 2 недели и 150 мг 1 раз в 2 недели), использованными в программе клинических исследований фазы III.
В контролируемых исследованиях 1158 пациентов (34,7%), получавшие препарат Пралуэнт, были в возрасте >65 лет, и 241 пациент (7,2%), получавшие препарат Пралуэнт, были в возрасте >75 лет. Не наблюдалось достоверных различий по безопасности и эффективности препарата по мере увеличения возраста пациентов.
Нежелательные реакции о которых сообщалось при применении препарата Пралуэнт в объединенных контролируемых исследованиях
Нежелательные реакции представлены в соответствии с частотой, классификация которой рекомендована Всемирной Организацией Здравоохранения: очень часто (>1/10), часто (от > 1/100 до <1/10), нечасто (от >1/1 000 до <1/100), редко (от >1/10 000 до <1/1 000), очень редко (<1/10000), частота неизвестна (частота не может быть определена на основании имеющихся данных).
Нарушения со стороны иммунной системы
Редко: гиперчувствительность, аллергический васкулит.
Нарушения со стороны дыхательной системы, органов грудной клетки и средостения
Часто: субъективные симптомы и объективные признаки со стороны верхних дыхательных путей, включая боль в ротоглотке, ринорею, чихание.
Нарушения со стороны кожи и подкожных тканей
Часто: кожный зуд.
Редко: крапивница, монетовидная экзема.
Общие расстройства и нарушения в месте введения
Часто: реакции в месте введения препарата, включая эритему/гиперемию, кожный зуд, отек, боль/болезненную чувствительность.
Описание отдельных нежелательных реакций
Реакции в месте введения препарата
Реакции в месте введения препарата, включая эритему/гиперемию, кожный зуд, отек, боль/болезненную чувствительность, отмечались у 6,1 % пациентов, получавших лечение алирокумабом, по сравнению с 4,1 % в контрольной группе. Большинство реакций в месте введения препарата были преходящими и слабо выраженными. Частота прекращения лечения вследствие развития реакций в месте введения была сопоставимой в обеих группах (0,2 % в группе алирокумаба по сравнению с 0,3 % в контрольной группе).
Генерализованные аллергические реакции
Генерализованные аллергические реакции чаще отмечались в группе алирокумаба, чем в контрольной группе, главным образом, с различием в частоте развития кожного зуда. Кожный зуд был обычно слабо выраженным и преходящим. Кроме этого, в контролируемых клинических исследованиях сообщалось о развитии редких и иногда серьезных аллергических реакций, таких как реакции гиперчувствительности, монетовидная экзема, крапивница и аллергический васкулит.
Значения ХС-ЛПНП <25 мг/дл
В объединенных контролируемых исследованиях у 796 пациентов из 3340 пациентов (23,8 %), получавших лечение препаратом Пралуэнт, отмечались два последовательно полученных значения концентрации ХС-ЛПНП в крови < 25 мг/дл, включая 288 пациентов (8,6 %) с двумя последовательно полученными значениями концентрации ХС-ЛПНП <15 мг/дл.
Эти случаи, главным образом, наблюдались, когда пациенты начинали и продолжали лечение препаратом Пралуэнт в дозе 150 мг 1 раз в 2 недели, независимо от исходных значений концентрации ХС-ЛПНП в крови или реакции на лечение.
Не было выявлено нежелательных реакций, связанных с этими значениями концентрации ХС-ЛПНП в крови.
Сердечно-сосудистые осложнения
В настоящее время продолжается исследование сердечно-сосудистых исходов, в котором главной конечной точкой являются подтвержденные экспертизой большие нежелательные сердечно-сосудистые осложнения (БНССО), такие как коронарная смерть, инфаркт миокарда, ишемический инсульт и нестабильная стенокардия, потребовавшая госпитализации.
При запланированном анализе объединенных исследований фазы III у 110 пациентов (3,5 %) в группе алирокумаба и у 53 пациентов (3 %) в контрольной группе (плацебо или активный контроль) наблюдались следующие, подтвержденные экспертизой, сердечно-сосудистые осложнения, развившиеся во время лечения: смерть, связанная с ишемической болезнью сердца, инфаркт миокарда, ишемический инсульт, нестабильная стенокардия, потребовавшая госпитализации, госпитализация по поводу хронической сердечной недостаточности и реваскуляризация. Подтвержденные БНССО наблюдались у 52 из 3182 пациентов (1,6 %) в группе алирокумаба и у 33 из 1792 пациентов (1,8%) в контрольной группе (плацебо и активный контроль).
При запланированном окончательном анализе клинического исследования LONG TERM подтвержденные экспертизой сердечно-сосудистые осложнения, развившиеся во время лечения, наблюдались у 72 из 1550 пациентов (4,6%) в группе алирокумаба и у 43 из 788 пациентов (5,1 %) в группе плацебо; подтвержденные экспертизой БНССО наблюдались у 27 из 1550 пациентов (1,7 %) в группе алирокумаба и у 26 из 788 пациентов (3,3 %) в группе плацебо. Отношения рисков (ОР) рассчитывали ретроспективно; для всех сердечно-сосудистых осложнений отношения рисков = 0,91 (95% ДИ, 0,62-1,34); для БНССО отношения рисков = 0,52 (95% ДИ, 0,31-0,90).
Смертность от всех причин
В клинических исследованиях фазы III смертность от всех причин составляла 0,6% (20 из 3182 пациентов) в группе алирокумаба и 0,9% (17 из 1792 пациентов) в контрольной группе. Главной причиной смертельных исходов были сердечно-сосудистые осложнения.
Иммуногенность/антитела к алирокумабу
Как и все другие протеины, применяющиеся для лечения, алирокумаб обладает потенциальной иммуногенностью. В исследованиях фазы III у 4,8 % пациентов, получавших лечение алирокумабом, отмечалось образование антител к алирокумабу (АА), по сравнению с 0,6 % в контрольной группе (плацебо и эзетимиб). У большинства этих пациентов наблюдались преходящие реакции образования АА с низкими титрами без нейтрализующей активности. По сравнению с пациентами, которые были АА-негативными, пациенты с АА-позитивным статусом не продемонстрировали различий в системной экспозиции алирокумаба, эффективности или безопасности, за исключением более высокой частоты развития реакций в месте введения препарата. Только у 1,2% пациентов в группе алирокумаба были выявлены нейтрализующие антитела. Большинство этих пациентов имело только один положительный результат анализа на наличие нейтрализующих антител; 10 пациентов (0,3%) имели два или более положительных результата анализа на наличие нейтрализующих антител. Данные у этих пациентов не подтверждают корреляции между наличием нейтрализующих антител и эффективностью в отношении снижения концентрации ХС-ЛПНП в крови и безопасностью.
Данные по иммуногенности зависят от чувствительности и специфичности методики их определения, а также от других факторов. Кроме этого, на наблюдаемую частоту АА-позитивного результата анализа оказывают влияние несколько факторов, включая обработку забранных проб крови, время забора крови, одновременно принимаемые лекарственные препараты и основное заболевание. По этим причинам сравнение частоты возникновения АА с частотой возникновения антител к другим препаратам может быть некорректным.
Передозировка
В контролируемых клинических исследованиях не было выявлено никаких изменений безопасности при более частом введении доз, чем рекомендованный режим дозирования 1 раз в 2 недели.
Взаимодействие с другими лекарственными средствами
Влияние алирокумаба на другие лекарственные средства
Так как алирокумаб является биологическим веществом, не ожидается каких-либо фармакокинетических эффектов алирокумаба на другие лекарственные препараты.
В клинических исследованиях при применении алирокумаба в комбинации с аторвастатином или розувастатином не наблюдалось каких-либо значимых изменений концентраций статинов в крови при повторных введениях алирокумаба, что указывает на то, что алирокумаб не влияет на изоферменты цитохрома Р450 (главным образом, изоферменты CYP3A4 и CYP2C9) и белки-транспортеры, такие как Р-гликопротеин (P-gp) и ОАТР (белок транспортер органических анионов).
Влияние других лекарственных средств на алирокумаб
Статины и другая липид-модифицирующая терапия, как известно, повышают синтез PCSK9, белка, являющегося мишенью алирокумаба. Повышение концентрации PCSK9 может привести к уменьшению системной экспозиции алирокумаба. Однако это не влияет на продолжительность действия препарата при применении алирокумаба 1 раз в 2 недели.
Особые указания
Аллергические реакции
В клинических исследованиях сообщалось о развитии генерализованных аллергических реакциях, включая зуд; также имелись сообщения о редких и иногда серьезных случаях аллергических реакций, таких как реакции гиперчувствительности, монетовидная экзема, крапивница и аллергический васкулит. При появлении симптомов и признаков серьезных аллергических реакций лечение препаратом Пралуэнт должно быть прекращено и следует начать проведение соответствующей симптоматической терапии.
Влияние на фертильность
Данные о неблагоприятном воздействии алирокумаба на фертильность отсутствуют.
Пожилые пациенты
Данные о применении алирокумаба у пациентов старше 75 лет ограничены. В контролируемых исследованиях 1158 пациентов (34,7%), получавших препарат Пралуэнт, были в возрасте >65 лет, и 241 пациент (7,2 %), получавших препарат Пралуэнт, были в возрасте >75 лет. Значимых различий в безопасности и эффективности препарата Пралуэнт с увеличением возраста не наблюдалось.
Почечная недостаточность
В клинических исследованиях количество пациентов с почечной недостаточностью тяжелой степени (клиренс креатинина <30 мл/мин/1,73 м2) было ограничено. Препарат Пралуэнт следует применять с осторожностью у пациентов с почечной недостаточностью тяжелой степени (см. раздел «С осторожностью»).
Печеночная недостаточность
Исследований алирокумаба у пациентов с печеночной недостаточностью тяжелой степени (класс С по шкале Чайлда-Пью) не проводились. Препарат Пралуэнт следует применять с осторожностью у данной категории пациентов (см. раздел «С осторожностью»).
Влияние на способность управлять транспортными средствами и механизмами
Препарат Пралуэнт не влияет или почти не влияет на способность управлять транспортными средствами и работать с механизмами.
Форма выпуска
Раствор для подкожного введения 75 мг/мл и 150 мг/мл.
По 1 мл в одноразовый шприц из прозрачного стекла (тип I), снабженный несъемной иглой из нержавеющей стали, защищенной колпачком из мягкого полимера.
По 1 шприцу в пластиковый блистер с покрытием.
По 1 блистеру или по 2 соединенных блистера или по 3 пары соединенных блистеров с инструкцией по применению в картонную пачку.
По 1 шприцу в шприц-ручку. По 1, 2 или 6 шприц-ручек с инструкцией по применению в картонную пачку, снабженную фиксатором.
Описание предварительно заполненных одноразовых шприц-ручек и инструкция по их использованию
Описание предварительно заполненных одноразовых шприцев и инструкция по их использованию
Условия хранения
Хранить при температуре от 2°С до 8°С. Не замораживать.
Хранить в недоступном для детей месте.
Срок годности
2 года.
Не применять по истечении срока годности, указанного на упаковке.
Условия отпуска
Отпускают по рецепту.
Производитель
Санофи Винтроп Индустрия, Франция (шприцы).
Юридическое лино, на имя которого выдано регистрационное удостоверение
АО Санофи-авентис груп, Франция.
Фасовщик (первичная упаковка)
Санофи Винтроп Индустрия, Франция (шприцы).
Упаковщик (вторичная (потребительская) упаковка) Санофи Винтроп Индустрия, Франция (шприцы).
Санофи-Авентис Дойчланд ГмбХ, Германия (шприц-ручки).
ЗАО «Санофи-Авентис Восток», Россия (шприц-ручки)
Выпускающий контроль качества
Санофи Винтроп Индустрия, Франция (шприцы).
Sanofi Winthrop Industrie, France.
1051 boulevard Industriel 76580 Le Trait, France.
Санофи-Авентис Дойчланд ГмбХ, Германия (шприц-ручки).
Sanofi-Aventis Deutschland GmbH, Germany.
Brueningstrasse 50/Industriepark Hoechst H500, H590, H600 65926 Frankfort am Main, Germany.
ЗАО «Санофи-Авентис Восток», Россия (шприц-ручки).
302516, Россия, Орловская область, Орловский район, с/п Болынекуликовское, ул. Ливенская, д. 1.
Претензии потребителей направлять по адресу в России:
Представительство АО «Санофи-авентис труп». 125009, г. Москва, ул. Тверская, 22.
В случае упаковки препарата на ЗАО «Санофи-Авентис Восток», Россия претензии потребителей направлять по адресу:
302516, Россия, Орловская область, Орловский район, с/п Болынекуликовское, ул. Ливенская, д. 1.
Купить Пралуэнт в Планета Здоровья
Купить Пралуэнт в ГорЗдрав
*Цены в Москве. Точная цена в Вашем городе будет указана на сайте аптеки.
Комментарии
(видны только специалистам, верифицированным редакцией МЕДИ РУ)

17.10.2023

Описание препарата Пралуэнт (раствор для подкожного введения, 75 мг/мл) основано на официальной инструкции, утверждено компанией-производителем в 2023 году
Дата согласования: 17.10.2023
Особые отметки:
![]()
![]()
Содержание
- Действующее вещество
- ATX
- Фармакологическая группа
- Нозологическая классификация (МКБ-10)
- Состав
- Описание лекарственной формы
- Фармакологическое действие
- Фармакодинамика
- Фармакокинетика
- Показания
- Противопоказания
- Применение при беременности и кормлении грудью
- Способ применения и дозы
- Побочные действия
- Взаимодействие
- Передозировка
- Особые указания
- Форма выпуска
- Производитель
- Условия отпуска из аптек
- Условия хранения
- Срок годности
- Заказ в аптеках Москвы
Действующее вещество
ATX
Фармакологическая группа
Состав
| Раствор для подкожного введения (в предварительно заполненных шприцах/шприц-ручках) | 1 мл |
| активное вещество: | |
| алирокумаб | 75/150 мг |
| вспомогательные вещества (полный перечень): L-гистидин и L-гистидина гидрохлорида моногидрат; сахароза; полисорбат 20; вода для инъекций |
Описание лекарственной формы
Прозрачная или слегка опалесцирующая, бесцветная или желтоватого цвета жидкость в предварительно заполненном шприце или предварительно заполненной шприц-ручке.
Фармакологическое действие
Фармакологическое действие
—
гиполипидемическое.
Фармакодинамика
Алирокумаб является полностью человеческим моноклональным антителом (изотип IgG1), мишенью которого является фермент пропротеиновая конвертаза субтилизин-кексин типа 9 (PCSK9). Алирокумаб производится с помощью технологии рекомбинантной ДНК с использованием суспензионной культуры клеток яичника китайского хомячка.
Алирокумаб имеет молекулярную массу приблизительно 146 кДа.
Механизм действия. PCSK9 связывается с рецепторами ЛПНП (Р-ЛПНП) на поверхности гепатоцитов, способствуя деградации Р-ЛПНП в печени. Р-ЛПНП являются главными рецепторами, которые выводят из системного кровотока циркулирующие ЛПНП, поэтому уменьшение количества Р-ЛПНП при связывании их с PCSK9 приводит к повышению концентрации Хс-ЛПНП в крови. Ингибируя связывание PCSK9 с Р-ЛПНП, алирокумаб увеличивает количество Р-ЛПНП для выведения ЛПНП, снижая таким образом концентрации Хс-ЛПНП в крови.
Р-ЛПНП также связывают богатые триглицеридами (ТГ) ремнантные ЛПОНП и липопротеины промежуточной плотности (ЛППП). Поэтому лечение алирокумабом может снижать концентрации этих ремнантных липопротеинов, о чем свидетельствует снижение аполипопротеина В (апо В), Хс липопротеинов, не являющихся липопротеинами высокой плотности (Хс-ЛПнеВП) и ТГ. Алирокумаб также снижает концентрацию липопротеина А (ЛП(а), являющегося формой ЛПНП, которая связаны с апо А. Однако было показано, что Р-ЛПНП имеют низкую аффинность к ЛП(а), в связи с чем точный механизм, с помощью которого алирокумаб снижает ЛП(а), полностью не установлен.
Генетические исследования
В генетических исследованиях, проведенных у человека, были выявлены разновидности гена PCSK9 с мутациями потери или повышения функции. У пациентов с одним аллелем PCSK9 с мутацией потери функции отмечались более низкие концентрации Хс-ЛПНП, которые коррелировали со значительно более низкой частотой развития ИБС. У некоторых пациентов были выявлены мутации потери функции в двух аллелях, и у них отмечались очень низкие концентрации Хс-ЛПНП в крови с концентрациями в крови Хс-ЛПВП и ТГ в нормальном диапазоне. Наоборот, мутации повышения функции в гене PCSK9 были выявлены у пациентов с повышенными концентрациями Хс-ЛПНП в крови и клиническим диагнозом семейной гиперхолестеринемии.
Наблюдательный анализ показал, что без лечения концентрации Хс-ЛПНП в крови у пациентов с мутациями повышения функции в гене PCSK9 находились в диапазоне, подобном наблюдавшемуся у пациентов с более часто встречающимися мутациями, вызывающими гетерозиготную форму семейной гиперхолестеринемии (такими как мутации в гене Р-ЛПНП), демонстрируя центральную роль фермента PCSK9 в метаболизме Хс-ЛПНП и его концентрациях в крови. В многоцентровом двойном слепом плацебо-контролируемом исследовании (продолжительностью 14 нед) 13 пациентов с гетерозиготной формой семейной гиперхолестеринемии, связанной с мутацией повышения функции в гене PCSK9, были рандомизированы в 2 группы: группу, получающую алирокумаб в дозе 150 мг 1 раз в 2 нед, и группу, получающую плацебо.
Среднее значение концентрации Хс-ЛПНП в крови составляло 151,5 мг/дл. На 2-й нед лечения среднее значение снижения исходной концентрации Хс-ЛПНП в крови составило 62,5% в группе пациентов, получавших алирокумаб, по сравнению с 8,8% у пациентов, получавших плацебо. На 8-й нед лечения среднее значение снижения концентрации Хс-ЛПНП в крови от исходного значения у всех пациентов, получавших алирокумаб, составило 72,4%.
Фармакодинамические свойства
Алирокумаб является полностью человеческим моноклональным антителом, которое подавляет активность PCSK9 как в исследованиях in vitro, так и в системах моделей in vivo. Большое количество исследований, проведенных у человека и животных, продемонстрировали центральную роль, которую играют повышенные концентрации Хс-ЛПНП в крови в возникновении и прогрессировании атеросклероза. Другие липопротеины, содержащие апо В-100, особенно богатые триглицеридами ремнантные липопротеины (образовавшиеся из ЛПОНП и ЛППП) и ЛП(а), также считаются способствующими развитию атеросклероза. Однако проведенные до настоящего времени исследования не выявили независимого влияния снижения концентраций этих липопротеинов на сердечно-сосудистую заболеваемость и смертность.
В исследованиях in vitro алирокумаб не индуцировал антителозависимую клеточноопосредованную токсичность и комплементзависимую цитотоксичность (Fc-опосредованную эффекторную функцию), как в присутствии, так и в отсутствии PCSK9. У алирокумаба, связанного с PCSK9, не наблюдалось образования нерастворимых иммунных комплексов, способных связывать протеины комплемента.
Клиническая эффективность/клинические исследования при первичной гиперхолестеринемии и смешанной дислипидемии
Резюме клинических исследований III фазы с использованием режима дозирования 75 и/или 150 мг каждые 2 нед (Q2W). Эффективность алирокумаба была изучена в 10 исследованиях III фазы (5 плацебо-контролируемых и 5 эзетимиб-контролируемых исследований), включавших 5296 рандомизированных пациентов с гиперхолестеринемией (несемейной и гетерозиготной формой семейной) или смешанной гиперлипидемией, из них 3188 пациентов были рандомизированы для приема алирокумаба. Три из этих 10 исследований были проведены исключительно у пациентов с гетерозиготной формой семейной гиперхолестеринемии. Большинство пациентов принимали одновременно липидмодифицирующую терапию, состоящую из максимально переносимых доз статинов в сочетании или без сочетания с другими липидмодифицирующими видами лечения и имели высокий и очень высокий сердечно-сосудистый риск. Два исследования были проведены у пациентов, которые не получали одновременно статины, включая одно исследование у пациентов с документированной непереносимостью статинов.
Два исследования (LONG TERM и HIGH FH), включающие в общей сложности 2416 пациентов, были проведены только с дозой 150 мг 1 раз в 2 нед. Восемь исследований были проведены с дозой 75 мг 1 раз в 2 нед, и критерием для повышения дозы до 150 мг 1 раз в 2 нед на 12-й нед было недостижение пациентами целевого значения концентрации Хс-ЛПНП в крови, основанного на степени их сердечно-сосудистого риска на 8-й нед лечения.
Главной конечной точкой эффективности во всех клинических исследованиях III фазы было среднее значение снижения концентрации Хс-ЛПНП в крови на 24-й нед по сравнению с плацебо и эзетимибом. Все указанные исследования достигли своей главной конечной точки.
В целом применение алирокумаба приводило также к статистически значимо большему значению процента снижения концентраций общего Хс (ОХ), Хс-ЛПнеВП, апо B и ЛП(а) по сравнению с плацебо/эзетимибом, независимо от того получали или нет пациенты статины. Алирокумаб также снижал концентрации ТГ и увеличивал концентрации Хс-ЛПВП и апо А-1 по сравнению с плацебо.
Результаты по эффективности были единообразными у пациентов с гетерозиготной формой семейной гиперхолестеринемии, без гетерозиготной семейной гиперхолестеринемии, у пациентов со смешанной дислипидемией и пациентов с сахарным диабетом. Снижение концентраций Хс-ЛПНП в крови было единообразным, независимо от того, принимались или не принимались пациентами одновременно статины, и от доз последних.
После прекращения введения алирокумаба не наблюдалось синдрома отмены и концентрация Хс-ЛПНП в крови постепенно возвращалась к исходным показателям.
Результаты оценки влияния на сердечно-сосудистые события. В заранее запланированном объединенном анализе данных исследований III фазы у 110 пациентов (3,5%) в группе алирокумаба и у 53 пациентов (3%) в контрольной группе (плацебо или активный контроль) наблюдались следующие, подтвержденные экспертизой, сердечно-сосудистые события, развившиеся во время лечения: смерть, связанная с ИБС, инфаркт миокарда, ишемический инсульт, нестабильная стенокардия, потребовавшая госпитализации, госпитализация по поводу ХСН и реваскуляризация.
Подтвержденные большие нежелательные сердечно-сосудистые события (БНССС), включавшие смерть от ИБС, инфаркт миокарда, ишемический инсульт или нестабильную стенокардию, требующую госпитализации, наблюдались у 52 из 3182 пациентов (1,6%) в группе алирокумаба и у 33 из 1792 пациентов (1,8%) в контрольной группе (плацебо и активный контроль).
В заранее запланированном анализе исследования LONG TERM подтвержденные экспертизой сердечно-сосудистые события, развившиеся во время лечения, наблюдались у 72 из 1550 пациентов (4,6%) в группе алирокумаба и у 40 из 788 пациентов (5,1%) в группе плацебо; подтвержденные экспертизой БНССС наблюдались у 27 из 1550 пациентов (1,7%) в группе алирокумаба и у 26 из 788 пациентов (3,3%) в группе плацебо. Относительный риск (ОР) рассчитывали ретроспективно; для всех сердечно-сосудистых событий ОР = 0,91 (95% ДИ: 0,62–1,34); для БНССС ОР = 0,52 (95% ДИ: 0,31–0,9).
Смерть от всех причин. В клинических исследованиях III фазы смерть от всех причин составляла 0,6% (20 из 3182 пациентов) в группе алирокумаба и 0,9% (17 из 1792 пациентов) в контрольной группе. Главной причиной смертельных исходов были сердечно-сосудистые события.
Исследование CHOICE I с использованием режима дозирования 300 мг каждые 4 нед (Q4W). Исследование CHOICE I — многоцентровое двойное слепое плацебо-контролируемое 48-недельное исследование, которое включало 540 пациентов, получающих максимально переносимую дозу статина с или без другой липидмодифицирующей терапии, и 252 пациента, не получавших статины. Пациенты были случайным образом распределены в группы лечения алирокумабом в дозе 300 мг Q4W, алирокумабом в дозе 75 мг Q2W или плацебо. 71,6% пациентов относились к категориям высокого или очень высокого сердечно-сосудистого риска. Все пациенты не имели целевых значений Хс-ЛПНП при включении в исследование. В группах алирокумаба доза могла быть скорректирована до 150 мг Q2W на 12-й нед у пациентов с сохраняющимся уровнем Хс-ЛПНП ≥70 мг/дл или ≥100 мг/дл в зависимости от их степени сердечно-сосудистого риска или у пациентов, у которых не отмечалось уменьшение Хс-ЛПНП на 30% от исходного уровня.
В когорте пациентов, принимавших статины, средняя исходная концентрация Хс-ЛПНП составила 112,7 мг/дл. На 12-й нед среднее процентное изменение от исходных значений Хс-ЛПНП в группе алирокумаба 300 мг Q4W составило −55,3% по сравнению с +1,1% для плацебо, при этом 77,3% пациентов, получавших алирокумаб 300 мг Q4W, достигли концентрации Хс-ЛПНП <70 мг/дл по сравнению с 9,3% в группе плацебо.
Увеличение дозы алирокумаба до 150 мг Q2W на 12-й нед потребовалось у 19,3% пациентов этой когорты. На 24-й нед среднее процентное изменение от исходных значений Хс-ЛПНП в группе алирокумаба 300 мг Q4W/150 мг Q2W составило −58,8% по сравнению с −0,1% для плацебо.
В когорте пациентов, не получавших сопутствующей терапии статинами, средняя исходная концентрация Хс-ЛПНП составляла 142,1 мг/дл. На 12-й нед среднее процентное изменение Хс-ЛПНП от исходного значения в группе алирокумаба 300 мг Q4W составило −58,4% по сравнению с +0,3% для группы плацебо, при этом 65,2% пациентов, получавших алирокумаб 300 мг Q4W, достигли концентрации Хс-ЛПНП <70 мг/дл по сравнению с 2,8% в группе плацебо. Увеличение дозы алирокумаба до 150 мг Q2W на 12-й нед потребовалось у 14,7% пациентов этой когорты. На 24-й нед среднее процентное изменение от исходных значений Хс-ЛПНП в группе алирокумаба 300 мг Q4W/150 мг Q2W составило −52,7% по сравнению с −0,3% для группы плацебо. В обеих когортах пациентов различие по сравнению с плацебо было статистически значимым на 24-й нед для всех параметров липидного обмена, за исключением апо А-1 в подгруппе пациентов, принимавших статины.
Исследование ESCAPE (с участием пациентов, получающих ЛПНП-аферез)
Многоцентровое двойное слепое плацебо-контролируемое 18-недельное исследование включало 62 пациентов с гетерозиготной формой семейной гиперхолестеринемии (41 пациент в группе алирокумаба и 21 пациент в группе плацебо), которым проводился ЛПНП-аферез еженедельно или каждые 2 нед. С 1-й по 6-ю неделю пациенты получали лечение алирокумабом в дозе 150 мг каждые 2 нед или плацебо, им также проводился ЛПНП-аферез с фиксированной частотой в соответствии с принятой индивидуальной схемой афереза до включения в исследование. Решение о том, требуется ли пациенту проведение афереза или нет, принималось с 7-й по 18-ю неделю на основании эффекта лечения у каждого пациента. К 6-й неделе средний процент изменения концентрации ХС-ЛПНП относительно исходного значения составил −53,7% в группе применения алирокумаба по сравнению с 1,6% в группе применения плацебо (p <0,0001). С 7-й по 18-ю неделю было отмечено уменьшение стандартизованной частоты проведения афереза на 50% (p <0,0001) в группе алирокумаба по сравнению с группой плацебо. Лечение аферезом было отменено у 11 из 41 пациента (26,8%) в группе алирокумаба, и у 27 из 41 пациента (65,8%) удалось избежать проведения по меньшей мере половины процедур. В группе плацебо ни у одного из пациентов лечение аферезом не было отменено полностью, и лишь у 2 из 21 пациента (9,5%) удалось избежать проведения по меньшей мере половины процедур.
Клиническая эффективность и безопасность при профилактике сердечно-сосудистых событий
Исследование ODYSSEY OUTCOMES. Многоцентровое двойное слепое плацебо-контролируемое исследование, включавшее 18924 взрослых пациента (9462 пациента, получавших алирокумаб, 9462 пациента, получавших плацебо), которые перенесли острый коронарный синдром (34,6% — инфаркт миокарда с подъемом сегмента ST; 48,6% — инфаркт миокарда без подъема сегмента ST; 16,8% — нестабильная стенокардия) за 4–52 нед до рандомизации и получали либо высокоинтенсивную терапию статинами (аторвастатин 40 или 80 мг; или розувастатин 20 или 40 мг) ± другая липидмодифицирующая терапия, либо максимально переносимые дозы указанных статинов ± другая липидмодифицирующая терапия. Все пациенты были рандомизированы в соотношении 1:1 в группы алирокумаба в дозе 75 мг Q2W или плацебо Q2W. Начиная со 2-го мес у пациентов с уровнем Хс-ЛПНП ≥50 мг/дл (1,29 ммоль /л) дозу алирокумаба увеличивали до 150 мг Q2W. В дальнейшем, если у пациентов, получавших алирокумаб в дозе 150 мг Q2W, регистрировалось 2 последовательных значения Хс-ЛПНП ниже 25 мг/дл (0,65 ммоль/л), дозу препарата снижали до 75 мг Q2W. Пациенты, получавшие препарат в дозе 75 мг Q2W, у которых регистрировались два последовательных значения Хс-ЛПНП ниже 15 мг/дл (0,39 ммоль/л), переводились в заслепленном режиме на плацебо. Примерно у 2615 (27,7%) из 9451 пациента, получавших алирокумаб, потребовалась коррекция дозы до 150 мг Q2W. В дальнейшем у 805 (30,8%) из этих 2615 пациентов доза была снижена до 75 мг Q2W. В целом 730 (7,7%) из 9451 пациентов в группе алирокумаба перешли на плацебо. Средняя продолжительность наблюдения в исследовании составила 33 мес. При рандомизации большинство пациентов (88,8%) получали высокоинтенсивную терапию статинами ± другая липидмодифицирующая терапия.
Алирокумаб значимо снижал риск первичной комбинированной конечной точки — время до первого БНССС, включая смерть от ИБС, нефатальный инфаркт миокарда, фатальный и нефатальный ишемический инсульт или нестабильную стенокардию, потребовавшую госпитализации (ОР 0,85; 95% ДИ: 0,78–0,93; р=0,0003).
Кроме этого, алирокумаб значимо снижал риск следующих комбинированных конечных точек: большие коронарные события (смерть от ИБС или инфаркт миокарда); коронарные события (большие коронарные события, нестабильная стенокардия, требующая госпитализации, или процедура коронарной реваскуляризации, связанная с ишемией); сердечно-сосудистые события (сердечно-сосудистая смерть, любое нефатальное коронарное событие или нефатальный ишемический инсульт); а также комбинированной конечной точки, включающей смерть от всех причин, нефатальный инфаркт миокарда или нефатальный ишемический инсульт.
Терапия алирокумабом была ассоциирована со снижением риска смерти от всех причин (ОР 0,85; 95% ДИ: 0,73–0,98; р=0,0261 (не скорректировано для множественных сравнений).
Нейрокогнитивные функции. В 96-недельном рандомизированном двойном слепом плацебо-контролируемом исследовании оценивалось влияние алирокумаба на нейрокогнитивные функции у пациентов с гетерозиготной семейной гиперхолестеринемией или несемейной гиперхолестеринемией при высоком или очень высоком сердечно-сосудистом риске. В течение 96 нед лечения алирокумаб не оказывал влияния на нейрокогнитивные функции. Процент пациентов с нейрокогнитивными расстройствами был низким в группах лечения алирокумабом (1,3%) и сопоставим с плацебо (1,7%). Никаких проблем безопасности, связанных с нейрокогнитивной функцией, не наблюдалось у пациентов, получавших алирокумаб, у которых были два последовательных значения Хс-ЛПНП <25 мг/дл или <15 мг/дл в период лечения.
Дети. 48-недельное открытое исследование было проведено для оценки эффективности и безопасности алирокумаба 75 мг 1 раз в 2 недели (масса тела <50 кг) или 150 мг 1 раз в 2 нед (масса тела ≥50 кг) у 18 педиатрических пациентов (от 8 до 17 лет) с гомозиготной семейной гиперхолестеринемией в дополнение к фоновой терапии. Пациенты получали алирокумаб 75 или 150 мг 1 раз в 2 нед без корректировки дозы до 12-й нед. Средний исходный уровень Хс-ЛПНП составлял 373 мг/дл. Среднее процентное изменение от исходного уровня Хс-ЛПНП к 12-й нед составлял -4,1% (95% ДИ: от -23,1% до 14,9%) в популяции ITT (n=18) и был связан с высокой вариабельностью реакции на снижение Хс-ЛПНП.
Фармакокинетика
Абсорбция. После п/к введения препарата Пралуэнт в дозе от 50 до 300 мг среднее время достижения Cmax алирокумаба в сыворотке крови составляло 3–7 дней.
Фармакокинетика алирокумаба после однократного п/к введения в дозе 75 мг в область живота, бедра или плеча была подобной. По данным популяционного анализа фармакокинетических показателей, абсолютная биодоступность алирокумаба после п/к введения составляла 85%.
Незначительно большее (2,1–2,7-кратное), чем пропорциональное дозе, увеличение концентраций алирокумаба наблюдалось при двукратном увеличении дозы с 75 до 150 мг 1 раз в 2 нед.
Равновесное состояние достигалось после введения 2–3 доз с двукратным коэффициентом накопления.
Месячная экспозиция алирокумаба при применении в дозе 300 мг каждые 4 нед аналогична таковой при применении дозы 150 мг каждые 2 нед.
Распределение. После в/в введения Vd алирокумаба составлял 0,04–0,05 л/кг, что указывает на распределение алирокумаба главным образом в системе кровообращения.
Биотрансформация. Специальные исследования метаболизма не проводились, т.к. алирокумаб является белком. Предполагается, что алирокумаб расщепляется на небольшие пептиды и отдельные аминокислоты.
Элиминация. У алирокумаба наблюдались 2 фазы выведения. В низких концентрациях элиминация происходит преимущественно через насыщаемую связь с мишенью (PCSK9), в то время как при более высоких концентрациях элиминация алирокумаба происходит преимущественно через ненасыщаемый протеолитический путь. По данным популяционного анализа фармакокинетических показателей, средний T1/2 алирокумаба составлял 17–20 дней у пациентов, получавших алирокумаб в монотерапии п/к в дозах 75 мг 1 раз в 2 нед или 150 мг 1 раз в 2 нед. При одновременном применении со статинами средний T1/2 алирокумаба составлял 12 дней.
Особые группы пациентов
Пожилой возраст. По данным популяционного анализа фармакокинетических показателей, возраст ассоциировался с небольшим различием в системной экспозиции алирокумаба в равновесном состоянии без влияния на его эффективность и безопасность.
Пол. По данным популяционного анализа фармакокинетических показателей, половая принадлежность пациентов не влияла на фармакокинетику алирокумаба.
Расовая принадлежность. По данным популяционного анализа фармакокинетических показателей, расовая принадлежность не оказывала влияния на фармакокинетику алирокумаба. После однократного п/к введения алирокумаба в дозах 100–300 мг не наблюдалось значимых различий в системной экспозиции у здоровых добровольцев, являющихся японцами и представителями белой расы.
Масса тела. По данным популяционного анализа фармакокинетических показателей, масса тела оказывала незначительное влияние на системную экспозицию алирокумаба без влияния на его эффективность и безопасность.
Нарушения функции печени. В клиническом исследовании I фазы при однократном п/к введении алирокумаба в дозе 75 мг фармакокинетические показатели алирокумаба у пациентов с печеночной недостаточностью легкой и средней степени тяжести были аналогичными таковым у пациентов с нормальной функцией печени. Доступные данные по фармакокинетике алирокумаба у пациентов с печеночной недостаточностью тяжелой степени отсутствуют.
Нарушения функции почек. Так как нет сведений о том, что моноклональные антитела выводятся почками, не ожидается, что функциональное состояние почек повлияет на фармакокинетику алирокумаба.
Популяционный анализ фармакокинетических показателей продемонстрировал, что нарушение функции почек легкой и средней степени тяжести не оказывало значимого воздействия на фармакокинетику алирокумаба. Данные по фармакокинетике алирокумаба у пациентов с тяжелыми нарушениями функции почек ограничены.
Дети. Доступны ограниченные фармакокинетические данные у 18 педиатрических пациентов (от 8 до 17 лет) с гомозиготной семейной гиперхолестеринемией. Достигнута равновесная средняя концентрация Ctrough алирокумаба на 12-й нед или ранее в группах, получающих алирокумаб в дозах 75 и 150 мг 1 раз в 2 нед. У детей младше 8 лет иследования не проводились.
Показания
Первичная гиперхолестеринемия и смешанная дислипидемия
Препарат Пралуэнт показан взрослым пациентам для лечения первичной гиперхолестеринемии (несемейной гиперхолестеринемии и гетерозиготной формы семейной гиперхолестеринемии) или смешанной дислипидемии, включая пациентов с сахарным диабетом типа 2, в дополнение к диете, для снижения концентрации Хс-ЛПНП, общего Хс, Хс-ЛПнеВП, Aпо B, ТГ и ЛПa и повышения концентраций ХС-ЛПВП и Апо А-1:
— в комбинации со статинами (ингибиторами ГМГ-КоА-редуктазы ) в сочетании или без сочетания с другой липидмодифицирующей терапией при невозможности достижения у пациентов целевой концентрации Хс-ЛПНП при приеме максимально переносимой дозы статинов;
— в монотерапии или как дополнение к другой, не относящейся к статинам липидмодифицирующей терапии, у пациентов с непереносимостью статинов или при наличии противопоказаний к их применению;
— для уменьшения частоты проведения ЛПНП-афереза у пациентов с гетерозиготной формой семейной гиперхолестеринемии.
Установленное атеросклеротическое сердечно-сосудистое заболевание
Препарат Пралуэнт показан взрослым пациентам с установленным атеросклеротическим сердечно-сосудистым заболеванием с целью снижения риска развития сердечно-сосудистых событий посредством снижения Хс-ЛПНП как дополнение к коррекции других факторов риска:
— в комбинации с максимально переносимыми дозами статинов в сочетании или без сочетания с другой липидмодифицирующей терапией;
— в монотерапии или как дополнение к другой, не относящейся к статинам, липидмодифицирующей терапии, у пациентов с непереносимостью статинов или при наличии противопоказаний к их применению.
Противопоказания
- гиперчувствительность к алирокумабу или какому-либо вспомогательному веществу препарата;
- беременность (эффективность и безопасность не установлены);
- период грудного вскармливания (эффективность и безопасность не установлены).
Применение при беременности и кормлении грудью
Беременность. Отсутствуют данные по применению препарата Пралуэнт у беременных женщин. Ожидается, что алирокумаб, как и другие антитела класса IgG, проникает через плацентарный барьер. Во время беременности применение препарата Пралуэнт не рекомендуется.
Период грудного вскармливания. Неизвестно, проникает ли алирокумаб в грудное молоко у человека. В связи с тем, что многие лекарственные препараты, и в т.ч. иммуноглобулины, проникают в грудное молоко у женщин, применение препарата Пралуэнт у женщин в период грудного вскармливания не рекомендуется. При необходимости применения препарата Пралуэнт в данный период следует прекратить грудное вскармливание.
Фертильность. Данные о неблагоприятном воздействии алирокумаба на фертильность отсутствуют.
Способ применения и дозы
Реклама: ООО «РЛС-Библиомед», ИНН 7714758963, erid=4CQwVszH9pUmKjt23pm
До начала терапии препаратом Пралуэнт необходимо исключить вторичные причины гиперлипидемиии или смешанной дислипидемии (например, нефротический синдром, гипотиреоз).
Препарат вводят подкожно.
Начальная доза препарата Пралуэнт составляет 75 мг 1 раз каждые 2 нед. У пациентов, которым требуется большее снижение концентрации Хс-ЛПНП (>60%), начальная доза препарата Пралуэнт может составлять 150 мг, которую также вводят 1 раз каждые 2 нед или 300 мг 1 раз каждые 4 нед (ежемесячно).
Дозу препарата Пралуэнт следует подбирать индивидуально на основании таких параметров, как исходные значения Хс-ЛПНП, цели терапии и ответ пациента на лечение. Концентрации липидов в крови можно оценивать через 4–8 нед после начала лечения или титрования дозы и проводить соответствующую коррекцию дозы. При необходимости дополнительного снижения концентрации Хс-ЛПНП у пациентов, которым препарат Пралуэнт назначался в дозе 75 мг 1 раз каждые 2 нед или 300 мг 1 раз каждые 4 нед, доза может быть скорректирована до максимальной дозы 150 мг 1 раз каждые 2 нед.
В случае пропуска очередного введения препарата пациенту необходимо как можно скорее ввести пропущенную дозу и затем продолжить терапию в соответствии с исходным режимом дозирования.
Пациенты, которым проводится ЛПНП-аферез
Препарат Пралуэнт может применяться у пациентов, которым проводится ЛПНП-аферез, независимо от времени и частоты проведения процедуры. Рекомендуемая доза — 150 мг каждые 2 нед. Концентрации Хс-ЛПНП можно оценивать до афереза для определения необходимости его проведения.
Особые группы пациентов
Пациенты пожилого возраста. У пациентов пожилого возраста коррекции дозы препарата Пралуэнт не требуется (см. «Особые указания»).
Печеночная недостаточность. У пациентов с печеночной недостаточностью легкой или средней степени тяжести коррекции дозы препарата Пралуэнт не требуется. Данные о применении препарата Пралуэнт у пациентов с печеночной недостаточностью тяжелой степени отсутствуют.
Почечная недостаточность. У пациентов с почечной недостаточностью легкой или средней степени тяжести коррекции дозы препарата Пралуэнт не требуется. Данные о применении препарата у пациентов с почечной недостаточностью тяжелой степени ограничены.
Масса тела. Не требуется коррекции режима дозирования в зависимости от массы тела пациентов.
Дети. Безопасность и эффективность применения препарата Пралуэнт у детей в возрасте до 18 лет не установлены.
Применение у детей младше 8 лет не изучалось.
Правила введения препарата
Препарат Пралуэнт применяют в виде подкожных инъекций, проводимых в области бедра, живота или плеча, с помощью одноразовой предварительно заполненной шприц-ручки или одноразового предварительно заполненного шприца.
При назначении дозы 300 мг препарат вводят либо в виде 1 инъекции по 300 мг, либо в виде 2 инъекций по 150 мг в разные места.
Рекомендуется менять места инъекций при проведении каждой инъекции.
Препарат Пралуэнт не следует вводить в области активных кожных заболеваний или повреждений, таких как солнечные ожоги, кожная сыпь, воспаления кожи или кожные инфекции.
Не следует вводить препарат Пралуэнт в то же место, в которое вводились другие лекарственные препараты.
Подготовка и обращение с препаратом
— пациент может вводить препарат Пралуэнт самостоятельно, или препарат может быть введен лицом, ухаживающим за пациентом, после предоставления медицинскими работниками информации по правильной технике проведения п/к инъекций;
— препараты для парентерального введения должны осматриваться визуально перед введением на предмет наличия видимых частиц и изменения цвета. Если раствор имеет измененный цвет или видимые частицы, его не следует использовать;
— перед использованием предварительно заполненных шприц-ручек следует согреть их до комнатной температуры;
— при необходимости (например, во время путешествия) препарат Пралуэнт можно хранить при температуре не выше 25 °C не более 30 дней. Защищать от света. После извлечения из холодильника препарат Пралуэнт должен быть использован в течение 30 дней или утилизирован;
— после использования шприц-ручки следует поместить ее в резистентный к проколам контейнер и утилизировать согласно местным (государственным) требованиям;
— не использовать контейнер повторно;
— всегда следует хранить контейнер в местах, недоступных для детей;
— пациенты и ухаживающие за ними лица должны получить руководство по надлежащей технике п/к введения, включая сведения по асептике, и по тому, как правильно использовать предварительно заполненные шприц-ручки;
— пациентов и ухаживающих за ними лиц следует информировать о том, что перед первым введением препарата Пралуэнт они должны внимательно прочитать инструкцию по применению препарата;
— пациентов и ухаживающих за ними лиц следует предупредить, что запрещено повторное использование шприц-ручек, и они должны быть проинструктированы, как безопасно утилизировать их после использования.
Правила обращения с препаратом
Хранить при температуре от 2 до 8 °C. Не замораживать. Не подвергать воздействию высоких температур. При необходимости (например, во время путешествия) препарат Пралуэнт можно хранить при температуре не выше 25 °C не более 30 дней. Защищать от света. После извлечения из холодильника препарат Пралуэнт должен быть использован в течение 30 дней или утилизирован.
Описание предварительно заполненных одноразовых шприц-ручек и инструкция по их использованию
Пралуэнт, раствор для п/к введения 75 мг/мл в предварительно заполненной шприц-ручке по 1 мл.
Пралуэнт, раствор для п/к введения 150 мг/мл в предварительно заполненной шприц-ручке по 1 мл.
Пралуэнт, раствор для п/к введения 150 мг/мл в предварительно заполненной шприц-ручке по 2 мл.
Инструкция по применению
Пациента необходимо информировать о том, что он должен сохранять эту инструкцию, т.к. она может понадобиться ему снова. Если у пациента появятся вопросы, то ему следует обратиться к лечащему врачу или провизору, или позвонить по номеру, указанному в листке-вкладыше.
Части шприц-ручки указаны на рисунках.

Важная информация
— шприц-ручка предназначена только для одноразового использования;
— шприц-ручка с зеленой кнопкой: в 1 мл раствора содержится 75 мг алирокумаба;
— шприц-ручка с серой кнопкой: в 1 мл раствора содержится 150 мг алирокумаба;
— препарат вводится п/к либо самим пациентом, либо другим лицом, ухаживающим за ним;
— шприц-ручку можно использовать только для одноразовой инъекции, и она должна быть утилизирована после применения.
Правила использования
— шприц-ручку следует хранить в недоступном для детей месте;
— перед использованием шприц-ручки следует внимательно прочитать инструкцию;
— необходимо следовать всем указаниям, представленным в инструкции, при каждом использовании шприц-ручек;
— неиспользованные шприц-ручки следует хранить в холодильнике при температуре от 2 до 8 °C.
Запрещено
— трогать желтый защитный колпачок;
— использовать шприц-ручку при протекании или повреждении;
— использовать шприц-ручку, если отсутствует голубой колпачок или он ненадежно закреплен;
— использовать шприц-ручку повторно;
— трясти шприц-ручку;
— замораживать шприц-ручку;
— подвергать шприц-ручку воздействию прямых солнечных лучей.
Этап А. Подготовка к введению
Перед введением препарата пациенту понадобятся:
— шприц-ручка с препаратом Пралуэнт;
— салфетки, смоченные спиртом;
— ватные тампоны или марля;
— контейнер, резистентный к проколам.
1. Проверить этикетку на шприц-ручке:
— убедиться, что взят правильный (нужный пациенту) препарат и правильная (нужная пациенту) доза (шприц-ручка с зеленой кнопкой — для дозы 75 мг/мл и шприц-ручка с серой кнопкой — для дозы 150 мг/мл);
— проверить срок годности. Не использовать по истечении срока годности.
2. Проверить окно:
— проверить, чтобы видимая в окно жидкость была прозрачной, бесцветной или бледно-желтой, без включений. Если имеются изменения цвета и видимые включения, то не следует использовать шприц-ручку (см. рисунок А);
— пациент может увидеть пузырьки воздуха. Это нормальное явление, не влияет на дальнейшее использование препарата;
— не использовать шприц-ручку, если окно желтого цвета и не содержит жидкости (см. рисунок B).
3. Оставить шприц-ручку согреваться при комнатной температуре в течение 30–40 мин:
— не нагревать шприц-ручку, она должна нагреться сама;
— использовать шприц-ручку как можно скорее после того, как она нагрелась;
— не класть шприц-ручку обратно в холодильник.
4. Подготовка места введения препарата:
— вымыть руки водой с мылом и вытереть их полотенцем;
— можно вводить препарат в область бедра, живота (за исключением области 5 см вокруг пупка), наружную поверхность плеча (см. рисунок);
— пациент может стоять или сидеть во время введения препарата;
— продезинфицировать место введения салфеткой, смоченной спиртом;
— не вводить препарат в болезненную, уплотненную, покрасневшую или воспаленную кожу;
— не вводить препарат в места рядом с видимыми венами;
— использовать разные места при каждом введении препарата;
— не вводить препарат в одно и то же место с другими препаратами.
Этап B. Как вводить препарат
1. После завершения всех пунктов «Этап А. Подготовка к введению» снять голубой колпачок:
— не снимать колпачок до тех пор, пока пациент не будет готов к введению препарата;
— не надевать голубой колпачок обратно.
2. Держать шприц-ручку, как показано на рисунке:
— не трогать желтый защитный колпачок;
— убедиться, что видно окно.
3. Прижать желтый защитный колпачок к коже примерно под углом 90°:
— прижимать шприц-ручку к коже и удерживать ее так, чтобы желтый защитный колпачок стал невидимым (вдавленным в кожу). Шприц-ручка не будет работать, если желтый защитный колпачок не вдавлен в кожу полностью;
— при необходимости следует зажать кожу, чтобы убедиться, что место введения плотное.
4. Нажать большим пальцем на зеленую (если используется шприц-ручка Пралуэнт, 75 мг) или серую кнопку (если используется шприц-ручка Пралуэнт, 150 мг) и сразу отпустить ее:
— будет слышен щелчок. Введение препарата началось;
— окно начнет желтеть.
5. Продолжать удерживать шприц-ручку плотно прижатой к коже после того, как отпущена кнопка:
— введение препарата может продолжаться до 20 с.
6. Проверить, что окно стало желтого цвета до того, как убрана шприц-ручка с кожи:
— не убирать шприц-ручку с кожи до тех пор, пока все окно не будет желтого цвета;
— введение препарата завершено, после того как все окно будет желтого цвета и будет слышен второй щелчок;
— если окно не стало полностью желтым, извлечь щприц-ручку и обратиться за помощью к лечащему врачу;
— не следует делать себе вторую инъекцию, не посоветовавшись с лечащим врачом.
7. Убрать шприц-ручку с кожи:
— не растирать кожу после введения препарата;
— если пациент видит кровь, необходимо прижать ватный тампон или марлю и удерживать их до тех пор, пока не остановится кровотечение.
8. Утилизировать шприц-ручку и колпачок:
— не надевать голубой колпачок повторно;
— поместить шприц-ручку в контейнер, резистентный к проколам;
— спросить у лечащего врача, как утилизировать контейнер;
— всегда хранить контейнер в недоступном для детей месте.
Пралуэнт, 150 мг/мл, раствор для подкожного введения в предварительно заполненной шприц-ручке по 2 мл
Инструкция по применению
Пациент должен быть информирован о том, что он должен сохранять эту инструкцию, т.к. она может понадобиться ему снова. Если у пациента появятся вопросы, то ему следует обратиться к лечащему врачу или провизору или позвонить по номеру телефона, указанному в листке-вкладыше. Части шприц-ручки показаны на рисунке.
Важная информация
— шприц-ручка предназначена только для одноразового использования; в 2 мл раствора содержится 300 мг алирокумаба.
— препарат вводится подкожно и может вводиться пациентом самостоятельно или лицом, ухаживающим за ним;
— перед использованием шприц-ручки пациент должен пройти обучение у лечащего врача;
— шприц-ручку можно использовать только для одноразовой инъекции, и она должна быть утилизирована после применения.
Правила использования
— шприц-ручку следует хранить в недоступном для детей и защищенном от света месте;
— перед использованием шприц-ручки следует внимательно прочитать инструкцию;
— необходимо следовать всем указаниям, представленным в инструкции, при каждом использовании шприц-ручки;
— неиспользованные шприц-ручки следует хранить в холодильнике при температуре от 2 до 8 °C.
Запрещено
— трогать желтый защитный колпачок;
— использовать шприц-ручку, если она протекает или повреждена;
— использовать шприц-ручку при отсутствии голубого колпачка, или если он ненадежно закреплен;
— использовать шприц-ручку повторно;
— трясти шприц-ручку;
— замораживать шприц-ручку;
— подвергать шприц-ручку воздействию тепла или прямых солнечных лучей.
Этап А. Подготовка к введению
Перед введением препарата пациенту понадобятся:
— шприц-ручка с препаратом Пралуэнт;
— салфетки, смоченные спиртом;
— ватные тампоны или марля;
— контейнер, резистентный к проколам.
1. Проверить этикетку на шприц-ручке:
— убедиться, что взят правильный (нужный пациенту) препарат и правильная (нужная пациенту) доза;
— проверить срок годности. Не использовать по истечении срока годности.
2. Проверить окно:
— проверить, чтобы видимая в окно жидкость была прозрачной, бесцветной или бледно-желтой, без включений. Если имеются изменения цвета и видимые включения, то не следует использовать шприц-ручку (см. рисунок А);
— пациент может увидеть пузырьки воздуха. Это нормальное явление, не влияет на дальнейшее использование препарата;
— не использовать шприц-ручку, если окно желтого цвета и не содержит жидкости (см. рисунок B).
3. Оставить шприц-ручку согреваться при комнатной температуре в течение 45 мин. Это важно для применения всей дозы и помогает минимизировать дискомфорт при введении препарата:
— не нагревать шприц-ручку, она должна нагреться сама;
— не класть шприц-ручку обратно в холодильник.
4. Подготовка места введения препарата:
— вымыть руки водой с мылом и вытереть их полотенцем;
— можно вводить препарат в область бедра, живота (за исключением области 5 см вокруг пупка), наружную поверхность плеча — может быть сделано только лицом, ухаживающим за пациентом (см. рисунок);
— пациент может стоять или сидеть во время введения препарата;
— продезинфицировать место введения салфеткой, смоченной спиртом;
— не вводить препарат в болезненную, уплотненную, покрасневшую или воспаленную кожу;
— не вводить препарат в места рядом с видимыми венами;
— использовать разные места при каждом введении препарата;
— не вводить препарат в одно и то же место с другими препаратами.
Этап B. Как вводить препарат
1. После завершения всех пунктов Этапа А. Подготовка к введению снять голубой колпачок:
— не снимать колпачок до тех пор, пока пациент не будет готов к введению препарата;
— не надевать голубой колпачок обратно;
— не использовать шприц-ручку при отсутствии голубого колпачка, или если он ненадежно закреплен.
2. Держать шприц-ручку, как показано на рисунке:
— не трогать желтый защитный колпачок;
— убедиться, что видно окно.
— не прижимать шприц-ручку к коже, пока осуществляющий инъекцию не будет готов к введению препарата.
3. Прижать желтый защитный колпачок к коже примерно под углом 90°:
— зажать кожу так, чтобы убедиться, что место введения плотное;
— прижимать шприц-ручку к коже до тех пор, пока желтый защитный колпачок не войдет в шприц-ручку до упора, и удерживать ее (см. рисунок выше);
— введение препарата не начнется, пока желтый защитный колпачок не будет полностью вжат;
— будет слышен щелчок. Введение препарата началось. Окно начнет желтеть.
4. Продолжать удерживать шприц-ручку, плотно прижатой к коже:
— возможен второй щелчок;
— убедиться, что окно стало желтым;
— затем медленно посчитать до 5.
5. Проверить, что окно стало желтого цвета до того, как будет убрана шприц-ручка от кожи:
— если окно не стало полностью желтым, извлечь шприц-ручку и обратиться за помощью к лечащему врачу;
— не делать себе вторую инъекцию, не посоветовавшись с лечащим врачом.
6. Убрать шприц-ручку от кожи:
— не растирать кожу после введения препарата;
— если пациент видит кровь, следует прижать ватный тампон или марлю и удерживать их до тех пор, пока не остановится кровотечение.
7. Утилизировать шприц-ручку и колпачок:
— не надевать голубой колпачок повторно;
— сразу же после введения препарата поместить шприц-ручку и колпачок в контейнер, устойчивый к проколам;
— спросить у лечащего врача, как утилизировать контейнер;
— всегда хранить контейнер в недоступном для детей месте.
Описание предварительно заполненных одноразовых шприцев и инструкция по их использованию
Пралуэнт, раствор для п/к введения 75 мг/мл в предварительно заполненном шприце.
Пралуэнт, раствор для п/к введения 150 мг/мл в предварительно заполненном шприце.
Инструкция по применению
Пациента необходимо информировать о том, что он должен сохранять эту инструкцию, т.к. она может понадобиться ему снова. Если у пациента появятся вопросы, то ему следует обратиться к лечащему врачу или провизору или позвонить по номеру, указанному в листке-вкладыше.
Части шприца показаны на рисунке.
Важная информация
Шприц предназначен для одноразового использования.
— шприц с зеленым поршнем: в 1 мл раствора содержится 75 мг алирокумаба;
— шприц с серым поршнем: в 1 мл раствора содержится 150 мг алирокумаба;
— препарат вводится п/к либо самим пациентом, либо другим лицом, ухаживающим за ним;
— шприц можно использовать только для однократной инъекции, и он должен быть утилизирован после использования.
Правила использования
— шприц следует хранить в недоступном для детей месте;
— перед использованием шприца следует внимательно прочитать инструкцию;
— необходимо следовать всем указаниям, представленным в инструкции, при каждом использовании шприца;
— неиспользованные шприцы следует хранить в холодильнике при температуре от 2 до 8 °C.
Запрещено
— трогать иглу;
— использовать шприц при протекании или повреждении;
— использовать шприц если отсутствует серый колпачок для иглы, или он ненадежно закреплен;
— использовать шприц повторно;
— трясти шприц;
— замораживать шприц;
— подвергать шприц воздействию прямых солнечных лучей.
Этап А. Подготовка к введению
Перед началом введения пациенту понадобятся:
— шприц с препаратом Пралуэнт;
— салфетки, смоченные спиртом;
— ватные тампоны или марля;
— контейнер, резистентный к проколам.
1. Перед началом следует:
— взять шприц из пакета, удерживая его за корпус.
2. Проверить этикетку на шприце:
— проверить, что взят правильный (нужный пациенту) препарат и правильная (нужная пациенту) доза (шприц с зеленым поршнем — для дозы 75 мг/мл и шприц с серым поршнем — для дозы 150 мг/мл);
— проверить срок годности, не использовать по истечении срока годности;
— проверить, чтобы жидкость в шприце была прозрачной, бесцветной или бледно-желтой, без включений. Если имеются изменения цвета и видимые включения, то не следует использовать шприц;
— проверить, что шприц не открыт и не поврежден.
3. Оставить шприц согреваться при комнатной температуре в течение 30–40 мин:
— не нагревать шприц, он должен нагреться сам;
— использовать шприц как можно скорее после того, как он нагреется;
— не класть шприц обратно в холодильник.
4. Подготовка места введения:
— вымыть руки водой и мылом и вытереть их полотенцем;
— можно вводить препарат в область бедра, живота (за исключением области 5 см вокруг пупка), наружную поверхность плеча (см. рисунок);
— пациент может стоять или сидеть во время введения препарата;
— продезинфицировать место введения салфеткой, смоченной спиртом;
— не вводить препарат в болезненную, уплотненную, покрасневшую или воспаленную кожу;
— не вводить препарат в места рядом с видимыми венами;
— использовать разные места при каждом введении препарата;
— не вводить препарат в одно и то же место с другими препаратами.
Этап B. Как вводить препарат
1. После завершения всех пунктов Этапа А. Подготовка к введению снять колпачок для иглы:
— не снимать колпачок до тех пор, пока пациент не будет готов к введению препарата;
— держать шприц за середину корпуса так, чтобы игла была направлена от пациента;
— не прикасаться к поршню;
— пациент может увидеть пузырьки воздуха. Это нормальное явление, не влияет на дальнейшее использование препарата. Не следует избавляться ни от каких пузырьков в шприце до начала инъекции;
— не надевать серый колпачок обратно.
2. При необходимости зажать кожу:
— большим и указательным пальцем зажать кожу так, чтобы получилась складка кожи в месте введения;
— удерживать кожную складку во время проведения всей инъекции.
3. Ввести иглу в складку кожи быстрым движением:
— использовать угол в 90° при сжатии 5 см кожи; использовать угол в 45° при сжатии 2 см кожи.
4. Опустить поршень вниз:
— ввести весь раствор медленным и равномерным нажатием на поршень;
5. Перед тем, как удалить иглу, следует убедиться, что шприц пуст:
— не удалять шприц, пока он полностью не опустеет;
— вытаскивать иглу из кожи следует под тем же углом, под каким она была введена;
— не растирать кожу после введения препарата;
— если видна кровь, необходимо прижать ватный тампон или марлю и удерживать их до тех пор, пока не остановится кровотечение.
6. Утилизировать шприц и колпачок:
— не надевать серый колпачок повторно;
— не использовать шприц повторно;
— поместить шприц в контейнер, резистентный к проколам;
— спросить у лечащего врача, как утилизировать контейнер;
— всегда хранить контейнер в недоступном для детей месте.
Подготовка и обращение с препаратом
— пациент может вводить препарат Пралуэнт самостоятельно, или препарат может быть введен лицом, ухаживающим за пациентом, после предоставления медицинскими работниками информации по правильной технике проведения п/к инъекций;
— препараты для парентерального введения должны осматриваться визуально перед введением на предмет наличия видимых частиц и изменения цвета. Если раствор имеет измененный цвет или видимые частицы, его не следует использовать;
— перед использованием предварительно заполненных шприцев следует нагреть их до комнатной температуры;
— при необходимости (например, во время путешествия) препарат Пралуэнт можно хранить при температуре не выше 25 °C не более 30 дней. Защищать от света. После извлечения из холодильника препарат Пралуэнт должен быть использован в течение 30 дней или утилизирован;
— после использования шприцы следует поместить в резистентный к проколам контейнер и утилизировать согласно местным (государственным) требованиям;
— не использовать контейнер повторно;
— всегда следует хранить контейнер в местах, недоступных для детей;
— пациенты и ухаживающие за ними лица должны получить руководство по надлежащей технике п/к введения, включая сведения по асептике, и по тому, как правильно использовать предварительно заполненные шприцы;
— пациентов и ухаживающих за ними лиц следует информировать о том, что перед первым введением препарата Пралуэнт они должны внимательно прочитать инструкцию по применению препарата;
— пациентов и ухаживающих за ними лиц следует предупредить, что запрещено повторное использование шприцев, и они должны быть проинструктированы, как безопасно утилизировать их после использования.
Побочные действия
Резюме профиля безопасности
Представленные ниже данные по безопасности отражают применение алирокумаба у 3340 пациентов, большинство из которых имели высокий или очень высокий риск развития сердечно-сосудистых заболеваний, получали алирокумаб в дозе 75 и/или 150 мг в виде п/к инъекций 1 раз в 2 нед с продолжительностью лечения до 18 мес (включая 2408 пациентов, получавших лечение алирокумабом в течение 52 нед, и 639 пациентов, получавших лечение алирокумабом в течение не менее 76 нед). Данные по безопасности основаны на объединенных результатах 9 плацебо-контролируемых исследований (4 исследования II фазы и 5 исследований III фазы (все исследования у пациентов, одновременно принимающих статины) и 5 контролируемых эзетимибом исследований III фазы (в 3 из которых пациенты одновременно принимали статины).
В 10 контролируемых исследованиях III фазы, в которые были включены пациенты с первичной гиперхолестеринемией и смешанной дислипидемией, наиболее частыми нежелательными реакциями (HP) (≥1% пациентов, получавших препарат Пралуэнт) были реакции в месте введения препарата, субъективные симптомы и объективные признаки со стороны верхних дыхательных путей и кожный зуд.
Наиболее частыми HP, приводящими к прекращению лечения у пациентов, получавших препарат Пралуэнт, были реакции в месте введения препарата.
Профиль безопасности препарата в исследовании ODYSSEY OUTCOMES (изучение сердечно-сосудистых исходов) в целом соответствовал данным, полученным в других контролируемых исследованиях III фазы.
Не наблюдалось различий в отношении профиля безопасности между двумя дозами (75 мг 1 раз в 2 нед и 150 мг 1 раз в 2 нед), использованными в программе клинических исследований III фазы.
HP, о которых сообщалось при применении препарата Пралуэнт в объединенных контролируемых исследованиях (включая исследование ODYSSEY OUTCOMES) и в ходе пострегистрационного применения препарата
Частота HP, выявленных в клинических исследованиях, определялась на основании новых случаев HP во всех клинических исследованиях III фазы. HP представлены по системно-органным классам. Категории частоты определялись исходя из следующих критериев: очень часто (≥1/10); часто (от ≥1/100 до <1/10); нечасто (от ≥1/1000 до <1/100); редко (от ≥1/10000 до <1/1000); очень редко (<1/10000); частота неизвестна (частота не может быть определена на основании имеющихся данных).
Частота HP, о которых сообщалось в период пострегистрационного применения препарата, основана на спонтанных сообщениях. Поэтому частота этих HP классифицируется как «неизвестна».
Со стороны иммунной системы: редко — реакции гиперчувствительности, аллергический васкулит.
Со стороны дыхательной системы, органов грудной клетки и средостения: часто — субъективные симптомы и объективные признаки со стороны верхних дыхательных путей, включая боль в ротоглотке, ринорею, чихание.
Со стороны кожи и подкожных тканей: часто — кожный зуд; редко — крапивница, монетовидная экзема; частота неизвестна — ангионевротический отек (пострегистрационные данные).
Общие расстройства и нарушения в месте введения: часто — реакции в месте введения препарата, включая эритему/гиперемию, кожный зуд, отек, боль/болезненную чувствительность; частота неизвестна — гриппоподобное состояние (пострегистрационные данные).
Описание отдельных HP
Реакции в месте введения препарата. Реакции в месте введения препарата, включая эритему/гиперемию, кожный зуд, отек, боль/болезненную чувствительность, отмечались у 6,1% пациентов, получавших лечение алирокумабом, по сравнению с 4,1% в контрольной группе. Большинство реакций в месте введения препарата были преходящими и слабо выраженными. Частота прекращения лечения вследствие развития реакций в месте введения была сопоставимой в обеих группах (0,2% в группе алирокумаба по сравнению с 0,3% в контрольной группе). В исследовании сердечно-сосудистых исходов (ODYSSEY OUTCOMES) реакции в месте введения препарата также наблюдались с большей частотой в группе пациентов, получавших алирокумаб, в сравнении с плацебо (3,8% в группе алирокумаба и 2,1% в группе плацебо).
Генерализованные аллергические реакции. Генерализованные аллергические реакции чаще отмечались в группе алирокумаба, чем в контрольной группе, главным образом с различием в частоте развития кожного зуда. Кожный зуд был обычно слабо выраженным и преходящим. Кроме этого, в контролируемых клинических исследованиях сообщалось о развитии редких и иногда серьезных аллергических реакций, таких как реакции гиперчувствительности, монетовидная экзема, крапивница и аллергический васкулит. В исследовании сердечно-сосудистых исходов (ODYSSEY OUTCOMES) генерализованные аллергические реакции наблюдались с одинаковой частотой в группах лечения, получавших алирокумаб и плацебо (7,9% в группе алирокумаба и 7,8% в группе плацебо).
Различий в частоте развития кожного зуда не наблюдалось.
Особые группы пациентов
Пожилые пациенты. В контролируемых исследованиях III фазы по первичной гиперхолестеринемии и смешанной дислипидемии, 1158 пациентов (34,7%), получавших препарат Пралуэнт, были в возрасте ≥65 лет, и 241 пациент (7,2%), получавших препарат Пралуэнт, были в возрасте ≥75 лет. В исследовании по сердечно-сосудистым исходам 2505 пациентов, получавших препарат Пралуэнт, были в возрасте ≥65 лет и 493 пациента, получавших препарат Пралуэнт, были в возрасте ≥75 лет. Не наблюдалось значимых различий по безопасности и эффективности препарата при увеличении возраста пациентов.
Дети. Опыт применения препарата Пралуэнт у детей ограничен 18 пациентами в возрасте от 8 до 17 лет с гомозиготной семейной гиперхолестеринемией. Новых результатов безопасности по сравнению с известным профилем безопасности для взрослых не наблюдалось.
Исследование с использованием режима дозирования 300 мг 1 раз каждые 4 нед
Профиль безопасности у пациентов, получавших режим дозирования 300 мг 1 раз в 4 нед, был аналогичен профилю безопасности с использованием режима дозирования каждые 2 нед, за исключением более высокой частоты развития реакций в местах инъекции. Реакции в месте инъекции препарата были зарегистрированы с частотой 16,6% в группе режима дозирования 300 мг 1 раз в 4 нед лечения и 7,9% в группе плацебо. Частота прекращения применения препарата из-за реакций в месте инъекции составляла 0,7% в группе режима дозирования 300 мг 1 раз в 4 нед лечения и 0% в группе плацебо.
Пациенты, которым проводится ЛПНП-аферез
Профиль безопасности у пациентов, которые получали лечение препаратом Пралуэнт в дозе 150 мг каждые 2 нед, и пациентов, которым проводился ЛПНП-аферез, был сходен с профилем безопасности, описанным в клинических исследованиях при использовании схемы с введением препарата каждые 2 нед у пациентов, которым не проводился ЛПНП-аферез.
Низкие значения Хс-ЛПНП — <25 мг/дл (0,65 ммоль/л). Во всех клинических исследованиях базовая гиполипидемическая терапия не могла быть скорректирована согласно дизайну исследования. Процент пациентов, достигших значений Хс-ЛПНП <25 мг/дл, (<0,65 ммоль/л) зависел как от исходного значения Хс-ЛПНП, так и от дозы алирокумаба.
В клинических и наблюдательных исследованиях препарата Пралуэнт не наблюдалось неблагоприятных последствий достижения очень низких значений Хс-ЛПНП.
Иммуногенность/антитела к алирокумабу
Как и все другие протеины, применяющиеся для лечения, алирокумаб обладает потенциальной иммуногенностью. В исследовании ODYSSEY OUTCOMES у 5,5% пациентов, получавших лечение алирокумабом в дозе 75 и/или 150 мг 1 раз в 2 нед, после начала лечения отмечалось образование антител к алирокумабу (АА) по сравнению с 1,6% в гpyппе пациентов, получавших плацебо. У большинства этих пациентов наблюдались преходящие реакции образования АА. Персистирующие АА наблюдались у 0,7% пациентов, получавших алирокумаб и у 0,4% пациентов, получавших плацебо. Нейтрализующие антитела наблюдались у 0,5% пациентов в группе алирокумаба и <0,1% в группе плацебо.
АА, включая нейтрализующие антитела, регистрировались в низких титрах и не имели клинически значимого влияния на эффективность и безопасность алирокумаба, за исключением более высокой частоты реакций в месте введения препарата у пациентов с АА в сравнении с пациентами без АА (7,5 по сравнению с 3,6%). Долгосрочные последствия продолжения лечения алирокумабом при наличии АА неизвестны.
В объединенных данных 10 исследований (включая исследования с плацебо-контролем и активным контролем), в которых пациенты получали лечение алирокумабом в дозах 75 и/или 150 мг 1 раз в 2 нед, а также в отдельных клинических исследованиях с режимом дозирования 75 мг 1 раз в 2 нед или 300 мг 1 раз в 4 нед (включая некоторых пациентов с коррекцией дозы до 150 мг 1 раз в 2 нед ), частота образования АА и нейтрализующих антител была сопоставима с той, которая наблюдалась в исследовании ODYSSEY OUTCOMES и описана выше.
Данные по иммуногенности зависят от чувствительности и специфичности методики их определения, а также от других факторов. Кроме этого, на наблюдаемую частоту АА-позитивного результата анализа оказывают влияние несколько факторов, включая обработку забранных проб крови, время забора крови, одновременно принимаемые лекарственные препараты и основное заболевание. По этим причинам сравнение частоты возникновения АА с частотой возникновения антител к другим препаратам может быть некорректным.
Сообщение о подозреваемых HP
Важно сообщать о подозреваемых HP после регистрации лекарственного препарата с целью обеспечения непрерывного мониторинга соотношения польза-риск лекарственного препарата. Медицинским работникам рекомендуется сообщать о любых подозреваемых нежелательных реакциях лекарственного препарата через национальные системы сообщения о HP государств — членов Евразийского экономического союза.
Российская Федерация. Федеральная Служба по надзору в cфере здравоохранения. 109074, Москва, Славянская пл., 4, стр. 1.
Тел.: (495) 698-45-38, (499) 578-02-30.
e-mail: pharm@roszdravnadzor.ru
www.roszdravnadzor.ru
Взаимодействие
Влияние алирокумаба на другие ЛС. Так как алирокумаб является биологическим веществом, не ожидается каких-либо фармакокинетических эффектов алирокумаба на другие лекарственные препараты.
В клинических исследованиях при применении алирокумаба в комбинации с аторвастатином или розувастатином не наблюдалось каких-либо значимых изменений концентраций статинов в крови при повторных введениях алирокумаба, что указывает на то, что алирокумаб не влияет на изоферменты цитохрома P450 (главным образом изоферменты CYP3A4 и CYP2C9) и белки-транспортеры, такие как P-gp и OATP (белок-транспортер органических анионов).
Влияние других ЛС на алирокумаб. Статины и другая липидмодифицирующая терапия, как известно, повышают синтез PCSK9-белка, являющегося мишенью алирокумаба. Это приводит к повышению мишень-опосредованного клиренса алирокумаба и снижению его системной экспозиции. Тем не менее снижение уровня Хс-ЛПНП сохранялось на протяжении терапии при применении алирокумаба 1 раз в 2 нед.
Передозировка
В контролируемых клинических исследованиях не было выявлено никаких изменений безопасности при более частом введении доз, чем рекомендованный режим дозирования 1 раз в 2 нед.
Лечение: не существует специального лечения при передозировке препарата Пралуэнт. В случае передозировки следует проводить симптоматическое лечение и поддерживающую терапию.
Особые указания
Аллергические реакции. В клинических исследованиях сообщалось о развитии генерализованных аллергических реакций, включая зуд; также имелись сообщения о редких и иногда серьезных случаях аллергических реакций, таких как реакции гиперчувствительности, монетовидная экзема, крапивница и аллергический васкулит. В ходе пострегистрационного применения препарата сообщалось о развитии ангионевротического отека (см. «Побочные действия») При появлении симптомов и признаков серьезных аллергических реакций лечение препаратом Пралуэнт следует прекратить и начать проведение соответствующей симптоматической терапии.
Пожилые пациенты. Данные о применении алирокумаба у пациентов >75 лет ограничены. В контролируемых исследованиях 1158 пациентов (34,7%), получавших препарат Пралуэнт, были в возрасте ≥65 лет, и 241 пациент (7,2%), получавший препарат Пралуэнт, был в возрасте ≥75 лет. Значимых различий в безопасности и эффективности препарата Пралуэнт с увеличением возраста не наблюдалось.
Почечная недостаточность. В клинических исследованиях количество пациентов с почечной недостаточностью тяжелой степени (Cl креатинина <30 мл/мин/1,73 м2) было ограничено. Препарат Пралуэнт следует применять с осторожностью у пациентов с почечной недостаточностью тяжелой степени.
Печеночная недостаточность. Исследования алирокумаба у пациентов с печеночной недостаточностью тяжелой степени (класс С по шкале Чайлд-Пью) не проводились. Препарат Пралуэнт следует применять с осторожностью у данной категории пациентов.
Влияние на способность управлять транспортными средствами и механизмами. Препарат Пралуэнт не влияет или почти не влияет на способность управлять транспортными средствами и работать с механизмами.
Форма выпуска
Раствор для подкожного введения, 75 мг/мл и 150 мг/мл.
Пралуэнт, 75 мг/мл, 150 мг/мл, в предварительно заполненных шприцах. По 1 мл в одноразовом шприце из прозрачного стекла (тип I) с уплотнителем из бромбутила и снабженном несъемной иглой из нержавеющей стали, защищенном колпачком из мягкого полимера. По 1 шприцу в пластиковый блистере с покрытием. По 1 блистеру или по 2 соединенных блистера или по 3 пары соединенных блистеров с листком-вкладышем помещают в картонную пачку.
Пралуэнт, 75 мг/мл, 150 мг/мл, в предварительно заполненных шприц-ручках. По 1 мл в одноразовом шприце из прозрачного стекла (тип I) с уплотнителем из бромбутила и снабженном несъемной иглой из нержавеющей стали, защищенной колпачком из мягкого полимера. По 1 шприцу помещают в одноразовую шприц-ручку. По 1, 2 или 6 шприц-ручек с листком-вкладышем помещают в картонную пачку, снабженную фиксатором.
Пралуэнт, 150 мг/мл, в предварительно заполненных шприц-ручках. По 2 мл в одноразовом шприце из прозрачного стекла (тип I) с уплотнителем из бромбутила и снабженном несъемной иглой из нержавеющей стали, защищенной колпачком из мягкого полимера. По 1 шприцу помещают в одноразовую шприц-ручку. По 1 или 3 шприц-ручки с листком-вкладышем помещают в картонную пачку, снабженную фиксатором.
Производитель
Держатель регистрационного удостоверения АО Санофи-авентис груп, Франция. 54, Рю де ля Боэти, 75008 Париж.
Тел.: +33 (0)1.53.77.40.00; факс: +33 (0)1.53.77.41.33
e-mail: www.sanofi.com
Представитель держателя регистрационного удостоверения/претензии потребителей направлять по адресу представительства АО «Санофи-авентис груп», Франция. РФ, 125009, Москва, ул. Тверская, 22.
Тел.: (495) 721-14-00.
e-mail: Sanofi.Russia@sanofi.com
Условия отпуска из аптек
По рецепту.
Условия хранения
При температуре 2–8 °C (не замораживать).
Хранить в недоступном для детей месте.
Срок годности
раствор для подкожного введения 75 мг/мл шприц — 2 года 6 мес.
Допускается хранение вне холодильника при температуре ниже 25 °C, использовать в течение 30 дней.
раствор для подкожного введения 150 мг/мл шприц — 2 года.
Допускается хранение вне холодильника при температуре ниже 25 °C, использовать в течение 30 дней.
Не применять по истечении срока годности, указанного на упаковке.
Представленная информация о ценах на препараты не является предложением о продаже или покупке товара.
Информация предназначена исключительно для сравнения цен в стационарных аптеках, осуществляющих деятельность в
соответствии со статьей 55 Федерального закона «Об обращении лекарственных средств» от 12.04.2010 № 61-ФЗ.
ОЛЕОКУПРИТ
- ОЛЕОКУПРИТ
- инсектофунгицид, предназначенный для ранневесенних обработок яблонь против зимующих стадий вредителей, парши и др. заболеваний. Эмульгирующийся концентрат, содержащий минер, нефтяное масло, 15% нафтенага меди и эмульгаторы. Применяется только ранней весной (в период набухания почек) при темп-ре воздуха не ниже + 4 оС концентрациях от 3,5 до 5% с расходом 40—60 кг концентрата на 1 га. Малотоксичен для теплокровных.
Сельско-хозяйственный энциклопедический словарь. — М.: Советская энциклопедия.
.
1989.
Смотреть что такое «ОЛЕОКУПРИТ» в других словарях:
-
олеокуприт — олеокуприт, инсектофунгицид, предназначенный для ранневесенних обработок яблонь против зимующих стадий вредителей, парши и других заболеваний. Эмульгирующийся концентрат, содержащий минеральное нефтяное масло, 15% нафтената меди и эмульгаторы.… … Сельское хозяйство. Большой энциклопедический словарь
-
НАФТЕНАТ МЕДИ — (купронафт, олеокуприт) Смесь медных солей нафтеновых кислот Вязкая маслянистая паста темно зеленого цвета. Хорошо растворима в нефтепродуктах и в ряде органических растворителей. С водой образует эмульсин. Выпускается в виде 50% й пасты,… … Пестициды и регуляторы роста растений
Клинико-фармакологическая группа
Ингибитор резорбции костной ткани. Моноклональное антитело
Действующее вещество
— денозумаб (denosumab)
Форма выпуска, состав и упаковка
Раствор для п/к введения в виде прозрачной или слегка опалесцирующей жидкости от бесцветного до слегка желтого цвета, практически свободной от видимых включений.
| 1 шприц (1 мл) | |
| деносумаб | 60 мг |
Вспомогательные вещества: уксусная кислота ледяная — 1 мг, натрия гидроксид — до pH 5.0-5.5, сорбитол (Е420) — 47 мг, полисорбат 20 — 0.1 мг, вода д/и — до 1 мл.
1 мл — шприцы предварительно заполненные одноразовые* (1) — лотки (1) — пачки картонные×.
1 мл — шприцы предварительно заполненные одноразовые* (1) — упаковки ячейковые контурные с термоэтикеткой (1) — пачки картонные×.
* с иглой 27G, колпачком и плунжером (с защитным устройством для иглы или без него).
× на каждую пачку наклеивают прозрачные защитные этикетки — контроль первого вскрытия, имеющие продольную цветную полосу.
Пачка картонная снабжена отрывной карточкой-напоминанием.
Препарат Пролиа представляет собой стерильный продукт и не содержит консервантов.
Фармакологическое действие
Деносумаб представляет собой полностью человеческое моноклональное антитело (IgG2), обладающее высокой аффинностью и специфичностью к лиганду рецептора активатора ядерного фактора каппа В (RANKL) и тем самым препятствует активации единственного рецептора RANKL — активатора ядерного фактора кВ (RANK), расположенного на поверхности остеокластов и их предшественников. Таким образом, предотвращение взаимодействия RANKL/RANK ингибирует образование, активацию и продолжительность существования остеокластов. В результате деносумаб уменьшает костную резорбцию и увеличивает массу и прочность кортикального и трабекулярного слоев кости.
Фармакодинамические эффекты
Назначение деносумаба в дозе 60 мг приводило к быстрому уменьшению сывороточных концентраций маркера резорбции костной ткани — 1С-телопептида (СТХ) — приблизительно на 70% в течение 6 ч после п/к введения и приблизительно на 85% в течение последующих 3 дней. Уменьшение концентрации СТХ оставалось стабильным в 6-месячном интервале между дозированием. Скорость снижения концентрации СТХ в сыворотке крови частично уменьшалась при снижении концентрации деносумаба в сыворотке крови, что отражает обратимость влияния деносумаба на ремоделирование кости. Данные эффекты наблюдались на протяжении всего курса лечения. Соответственно физиологической взаимосвязи процессов образования и резорбции при ремоделировании костной ткани наблюдалось уменьшение содержания маркеров образования кости (например, костно-специфической щелочной фосфатазы и сывороточного N-концевого пропептида коллагена 1 типа) с первого месяца после введения первой дозы деносумаба. Маркеры ремоделирования кости (маркеры образования кости и резорбции кости), как правило, достигали концентраций периода до начала лечения не позднее, чем через 9 месяцев после приема последней дозы препарата. После возобновления лечения деносумабом степень снижения концентраций СТХ была сходна со степенью снижения концентрации СТХ в начале курса лечения деносумабом.
Было показано, что перевод с лечения алендроновой кислотой (средняя продолжительность применения — 3 года) на деносумаб приводит к дополнительному снижению концентрации СТХ в сыворотке по сравнению с группой женщин в постменопаузе с низкой костной массой, продолжавших лечение алендроновой кислотой. В то же время изменения содержания кальция в сыворотке были одинаковыми в обеих группах.
В экспериментальных исследованиях ингибирование RANK/RANKL одновременно со связыванием остеопротегерина с Fc-фрагментом (OPG-Fc), приводило к замедлению роста кости и нарушению прорезывания зубов. Поэтому, лечение деносумабом может тормозить рост костей с открытыми зонами роста у детей и приводить к нарушениям прорезывания зубов.
Иммуногенность
Деносумаб — человеческое моноклональное антитело, поэтому, как и для других лекарственных средств белковой природы существует теоретический риск иммуногенности. Более чем 13 000 пациентов были обследованы на предмет образования, связывающих антител с использованием метода чувствительной электрохемилюминесценции в сочетании с иммунологическим анализом. Менее чем у 1% пациентов, принимавших деносумаб в течение 5 лет, определялись антитела (включая существовавшие ранее, транзиторные и растущие). Серопозитивные пациенты были далее обследованы на предмет образования нейтрализующих антител, используя хемилюминесцентный анализ в культуре клеток in vitro, нейтрализующих антител не обнаружено. Не было выявлено изменений фармакокинетического профиля, токсического профиля или клинического ответа, обусловленных образованием антител.
Клиническая эффективность
Лечение остеопороза в постменопаузе
У женщин с постменопаузным остеопорозом Пролиа увеличивает минеральную плотность кости (МПК), уменьшает частоту переломов шейки бедра, вертебральных и невертебральных переломов. Эффективность и безопасность деносумаба в лечении постменопаузного остеопороза была доказана в исследовании, длительностью 3 года. Результаты исследования показывают, что деносумаб существенно, в сравнении с плацебо снижает риск возникновения вертебральных и невертебральных переломов, переломов шейки бедра у женщин с остеопорозом в постменопаузе. В исследование было включено 7808 женщин, из которых у 23% отмечались часто встречающиеся переломы позвонков. Все три конечные точки эффективности в отношений переломов достигали статистически значимых значений, оцениваемых по предварительно заданной последовательной схеме тестирования.
Снижение риска возникновения новых вертебральных переломов при применении деносумаба в течение более чем 3 лет оставалось стабильным и значимым. Риск снижался независимо от 10-летней вероятности возникновения крупных остеопоротических переломов. На снижение риска также не влияли наличие часто встречающихся переломов позвонков в анамнезе, невертебральные переломы, возраст, пациентов, МПК, уровень ремоделирования кости и предшествующая терапия по поводу остеопороза.
У женщин старше 75 лет в постменопаузе деносумаб уменьшал частоту возникновения новых вертебральных переломов, и, по данным post hoc анализа, уменьшал частоту переломов шейки бедра.
Уменьшение частоты возникновения невертебральных переломов наблюдалось независимо от 10-летней вероятности возникновения крупных остеопоротических переломов.
Деносумаб существенно, по сравнению с плацебо, увеличивал МПК во всех анатомических областях. МПК определяли через 1 год, 2 и 3 года после начала терапии. Сходное влияние на МПК отмечено в поясничном отделе позвоночника независимо от возраста, расовой принадлежности, индекса массы тела (ИМТ), МПК и ремоделирования кости.
Гистологические исследования подтвердили нормальную архитектонику кости и, как и ожидалось, снижение костного ремоделирования по сравнению с плацебо. Не отмечено патологических изменений, включая фиброз, остеомаляцию и нарушение архитектоники костной ткани.
Клиническая эффективность при лечении потери костной массы, вызванной гормондепривационной терапией или терапией ингибиторами ароматазы
Лечение потери костной массы, вызванной депривацией андрогенов
Эффективность и безопасность деносумаба при лечении потери костной массы, ассоциированной со снижением концентрации андрогенов, были доказаны в 3-х летнем исследовании включавшем 1 468 пациентов с неметастатическим раком предстательной железы.
Существенное увеличение МПК определяли в поясничном отделе позвоночника, всей бедренной кости, шейке бедренной кости, вертеле бедренной кости спустя 1 месяц после приема первой дозы. Увеличение МПК в поясничном отделе позвоночника не зависело от возраста, расовой принадлежности, географического региона, ИМТ, начальных значений МПК, ремоделирования кости; продолжительности проведения гормондепривационной терапии и наличия вертебрального перелома в анамнезе.
Деносумаб значительно уменьшал риск возникновения новых вертебральных переломов на протяжении 3 лет применения. Уменьшение риска наблюдалось через 1 год и через 2 года после начала терапии. Деносумаб также снижал риск возникновения более чем одного остеопоротического перелома любой локализации.
Лечение потери костной массы, у женщин, получающих терапию ингибиторами ароматазы по поводу рака молочной железы
Эффективность и безопасность деносумаба в лечении потери костной массы, вызванной адъювантной терапией ингибитором ароматазы, оценивалась в 2-летнем исследовании, включавшем 252 пациентки с неметастатическим раком молочной железы. Деносумаб значительно увеличивал МПК во всех анатомических областях, по сравнению с плацебо, в течение 2 лет. Увеличение МПК наблюдалось в поясничном отделе позвоночника спустя месяц после приема первой дозы. Положительное влияние на МПК в люмбальном отделе позвоночника отмечали вне зависимости от возраста, продолжительности терапии ингибитором ароматазы, ИМТ, предшествующей химиотерапии, предшествующего использования селективного модулятора рецепторов эстрогена (СМРЭ) и времени, прошедшего от начала менопаузы.
Фармакокинетика
При п/к введении деносумаб характеризуется нелинейной фармакокинетикой, дозозависимой в широком диапазоне доз, и дозозависимым увеличением экспозиции для дозы в 60 мг (или 1 мг/кг) и выше.
Всасывание
После п/к введения деносумаба в дозе 60 мг биодоступность составила 61% и Cmax деносумаба — 6 мкг/мл (диапазон 1-17 мкг/мл), данные параметры наблюдались через 10 дней (диапазон 2-28 дней). После достижения Cmax содержание препарата в сыворотке крови снижалось с T1/2 26 дней (диапазон 6-52 дня) и далее в течение 3 месяцев (диапазон 1.5-4.5 месяцев). У 53% пациентов деносумаб не обнаруживался в сыворотке крови после 6 месяцев от последнего введения препарата.
Распределение
Не наблюдалось изменений фармакокинетических параметров деносумаба, а также кумуляции за все время приема многократных доз препарата по 60 мг каждые 6 месяцев
Метаболизм
Деносумаб состоит из аминокислот и углеводов, как обычный иммуноглобулин. На основании данных доклинических исследований, ожидается, что метаболизм деносумаба будет происходить по пути клиренса иммуноглобулинов, результатом которого будет распад на небольшие пептидные цепи и отдельные аминокислоты.
Выведение
На основании доклинических данных, выведение деносумаба будет происходить по пути выведения всех иммуноглобулинов, результатом которого будет распад на небольшие пептидные цепи и отдельные аминокислоты.
Отдельные группы пациентов
Возраст не оказывает значимого влияния на фармакокинетику деносумаба по данным фармакокинетического анализа в популяции пациентов от 28 лет до 87 лет.
Фармакокинетика у детей не изучалась.
Фармакокинетика деносумаба не зависит от расовой принадлежности.
В исследовании на 55 пациентах с различной степенью почечной недостаточности, включая пациентов, находящихся на диализе, степень почечной недостаточности не оказывала влияния на фармакокинетику и фармакодинамику деносумаба; поэтому не требуется коррекции режима дозирования деносумаба при хронической почечной недостаточности.
Исследований влияния недостаточности функции печени на фармакокинетику деносумаба не проводилось.
Показания
- лечение постменопаузного остеопороза;
- лечение потери костной массы у женщин, получающих терапию ингибиторами ароматазы по поводу рака молочной железы и у мужчин с раком предстательной железы, получающим гормон-депривационную терапию.
Противопоказания
- гипокальциемия;
- повышенная чувствительность к любому из компонентов препарата.
Дозировка
Введение
Проведение инъекции препарата требует предварительного обучения — см. рекомендации по введению препарата, приведенные в конце настоящей инструкции.
Рекомендуемая доза препарата Пролиа — одна п/к инъекция 60 мг каждые 6 месяцев. В течение курса лечения рекомендуется дополнительно принимать препараты кальция и витамин D.
Дети
Препарат Пролиа не рекомендован к применению в педиатрии, так как эффективность и безопасность данного препарата не изучались в этой возрастной группе.
Пациенты пожилого возраста
Основываясь на имеющихся данных об эффективности и безопасности препарата в данной возрастной группе, не требуется коррекции режима дозирования препарата.
Почечная недостаточность
Основываясь на имеющихся данных об эффективности и безопасности препарата в данной группе пациентов, не требуется коррекции режима дозирования препарата.
У пациентов с тяжелой почечной недостаточностью (клиренс креатинина <30 мл/мин) или находящихся на диализе существует большой риск развития гипокальциемии. Таким пациентам необходимо дополнительно принимать препараты кальция и витамин D.
Печеночная недостаточность
Эффективность и безопасность не изучались.
Инструкция по использованию
Следует оценить раствор перед введением на предмет наличия включений или изменения цвета. Раствор нельзя использовать при помутнении или изменении цвета. Не встряхивать.
Чтобы избежать дискомфорта в месте введения, следует согреть раствор до комнатной температуры (до 25°С) перед инъекцией, а затем медленно ввести все содержимое предварительно заполненного шприца. Шприц с остатками препарата выбросить.
Любые количества неиспользованного препарата или неиспользованные материалы должны быть уничтожены в соответствии с местными требованиями
Побочные действия
Данные, полученные при контролируемом применении в клинических исследованиях.
Нежелательные реакции приводятся по классам систем органов в терминах Медицинского словаря регуляторной деятельности (MedDRA).
Частота возникновения нежелательных явлений классифицировалась следующим образом: очень часто (≥1/10), часто (≥1/100, <1/10), иногда (≥1/1000, <1/100), редко (≥1/10 000, <1/1000), очень редко (<1/10 000), включая отдельные случаи.
В каждой группе систем органов и частоты сообщений нежелательные реакции приводятся по убыванию степени серьезности.
| Класс системы органов | Частота | Нежелательная реакция |
| Инфекции и инвазии | Нечасто | воспаление подкожной клетчатки |
| Со стороны метаболизма и электролитного обмена | Очень редко | гипокальциемия |
| Со стороны органа зрения | Часто | катаракта у мужчин, получающих андроген-депривационную терапию по поводу рака предстательной железы |
| Со стороны кожи и подкожно-жировой клетчатки | Нечасто | экзема, включая дерматиты, аллергические дерматиты, атопический дерматит, контактный дерматит |
| Со стороны костно-мышечной системы и соединительной ткани | Часто Редко |
Боль в конечностях Остеонекроз челюсти |
Передозировка
В клинических исследованиях не отмечено случаев передозировки препарата.
В клинических исследованиях вводили дозы деносумаба до 180 > мг каждые 4 недели (кумулятивная доза до 1080 мг в 6 месяцев).
Лекарственное взаимодействие
Исследований лекарственных взаимодействий не проводилось.
Препарат не следует смешивать с другими лекарственными средствами.
Особые указания
Рекомендуется прием препаратов кальция и витамина D во время применения препарата Пролиа.
Гипокальциемия может быть скорректирована приемом препаратов кальция и витамина D в адекватных дозах перед началом терапии деносумабом. Рекомендуется контроль концентрации кальция у пациентов, предрасположенных к гипокальциемии.
У пациентов, получающих препарат Пролиа, могут развиться инфекции кожи и ее придатков (преимущественно воспаление подкожной клетчатки), в отдельных случаях требующие госпитализации. О таких реакциях чаще сообщалось для группы деносумаба (0.4%) чем группы плацебо (0.1%). При этом общая частота возникновения кожных инфекций сравнима в группах деносумаба и плацебо. Пациентов следует проинструктировать незамедлительно обратиться за врачебной помощью в случае развития симптомов и признаков воспаления подкожной клетчатки.
У пациентов с распространенным раком, получавших 120 мг деносумаба каждые 4 недели сообщалось о развитии случаев остеонекроза челюсти. Имеются отдельные сообщения о развитии остеонекроза челюсти на дозе 60 мг каждые 6 месяцев.
Лица с аллергией на латекс не должны касаться резинового колпачка иглы (производное латекса).
Влияние на способность к управлению транспортными средствами и механизмами
Исследований влияния на способность к вождению автотранспортных средств и управлению механизмами не проводилось.
Беременность и лактация
Нет каких-либо данных по применению препарата во время беременности Пролиа не рекомендуется для применения у беременных женщин.
В токсикологических исследованиях на низших приматах было показано, что в дозах, 100-кратно превышающих рекомендуемые для клинического применения, деносумаб не оказывал влияния на фертильность или развитие плода.
Эксперименты на мышах с выключенным геном показали, что отсутствие RANKL может приводить к нарушению развития лимфатических узлов у плода, а в постнатальном периоде может быть причиной нарушения прорезывания зубов, и роста костей; также возможно влияние на созревание молочной железы, что может приводить к ослаблению лактации.
Неизвестно, выводится ли деносумаб в грудное молоко. Поскольку известно, что потенциально деносумаб может вызывать нежелательные реакции у детей грудного возраста, необходимо или прекратить грудное вскармливание, или отменить препарат.
Применение в детском возрасте
Препарат Пролиа не рекомендован к применению в педиатрии, так как эффективность и безопасность данного препарата не изучались в этой возрастной группе.
При нарушениях функции почек
Основываясь на имеющихся данных об эффективности и безопасности препарата в данной группе пациентов, не требуется коррекции режима дозирования препарата.
У пациентов с тяжелой почечной недостаточностью (клиренс креатинина <30 мл/мин) или находящихся на диализе существует большой риск развития гипокальциемии. Таким пациентам необходимо дополнительно принимать препараты кальция и витамин D.
При нарушениях функции печени
Эффективность и безопасность не изучались.
Применение в пожилом возрасте
Основываясь на имеющихся данных об эффективности и безопасности препарата в данной возрастной группе, не требуется коррекции режима дозирования препарата.
Условия отпуска из аптек
Препарат отпускается по рецепту.
Условия и сроки хранения
Хранить при температуре 2-8°С. Не замораживать. Хранить в оригинальной упаковке для защиты от света.
Не встряхивать. Хранить в недоступном для детей месте!
После изъятия из холодильника Пролиа может храниться при комнатной температуре не выше 25°С в оригинальной упаковке не более 30 дней.
Срок годности — 3 года.
Описание препарата ПРОЛИА основано на официально утвержденной инструкции по применению и утверждено компанией–производителем.
Предоставленная информация о ценах на препараты не является предложением о продаже или покупке товара. Информация предназначена исключительно для сравнения цен в стационарных аптеках, осуществляющих деятельность в соответствии со статьей 55 ФЗ «Об обращении лекарственных средств».
Обнаружили ошибку? Выделите ее и нажмите Ctrl+Enter.
Состав
Активные компоненты: сакубитрила и валсартана гидратный комплекс натриевых солей.
Одна таблетка 56,551/113,103/226,206 мг, в пересчете на безводную кислотную форму 50/100/200 мг – эквивалентно количеству сакубитрила 24,3/48,6/97,2 мг и количеству валсартана 25,7/51,4/102,8 мг.
Дополнительные компоненты: МКЦ, гипромеллоза, кросповидон, магния стеарат, тальк, кремния диоксид коллоидный.
Состав оболочки:
- премикс оболочки белый (гипромеллоза, титана диоксид, макрогол 4000, тальк);
- премикс оболочки красный (гипромеллоза, краситель железа оксид красный, макрогол 4000, тальк);
- премикс оболочки черный (гипромеллоза, краситель железа оксид черный (E172), макрогол 4000, тальк).
Форма выпуска
Таблетки, 50 мг, покрытые пленочной оболочкой, с фаской, овальной формы, без риски, двояковыпуклые, цвет белый с фиолетовым оттенком. Одна сторона имеет гравировку «LZ», другая – «NVR».
Таблетки 100 мг светло-желтого цвета.
Таблетки 200 мг светло-розового цвета.
Таблетки для приема внутрь, №14, 28, 56; 14 шт.; блистер, пачка картонная, листок-вкладыш с инструкцией.
Фармакологическое действие
Кардиопротективное, гипотензивное.
Фармакодинамика и фармакокинетика
Терапевтический эффект Valsartan + Sacubitril основан на ингибировании неприлизина с помощью метаболита Sacubitril и отключении рецепторов ангиотензина Valsartan (антагонисты рецепторов).
Воздействие сакубитрилата приводит к росту пептидов, нейтрализуемых неприлизином. Создаются синергические позитивные результаты, полученные от двух компонентов одновременно. В итоге достигается благоприятное воздействие на кровеносные сосуды, общее состояние сердечной мышцы и почек у страдающих коронарной недостаточностью пациентов.
Эффект возможен благодаря стимуляции деятельности рецепторов связанных клеточных мембран, координированных с гуанилатциклазой.
В результате наблюдается:
- значительное количество циклических гуанозинмонофосфатов (вызывает снятие спазма сосудов);
- мочегонный эффект и увеличение натрийуреза;
- уменьшение симпатической активности;
- изменение показателей при пробе Реберга;
- ускорение скорости тока крови в почках;
- влияние на процесс высвобождения почками гормона ренина.
После приема дозы препарата также наблюдается предотвращение стойкой активации РААС. Как известно, именно по этой причине происходит спазм кровеносных сосудов, скопление натрия и жидкости в почечных тканях, стимуляция процесса увеличения числа клеток благодаря митозу, а также последующие изменения структур и функций сердца и сосудов, ставшие негативным следствием многих патологий сосудов и сердечной мышцы.
Ингредиенты лекарственной формы также предотвращают дезадаптивное ремоделирование сердца и сосудов.
Фармакодинамический эффект приема комплексного препарата изучался у пациентов, страдающих тяжелой и длительно протекающей коронарной недостаточностью и у группы людей, не имеющих проблем со здоровьем. Сравнивался результат приема однократной дозы и эффект от длительной по времени схемы лечения. Зарегистрированный результат целиком и полностью соответствовал требованиям, предъявляемым к комплексам для снижения артериального давления.
Клинические испытания
Применение препарата в течение недели у испытуемых с зарегистрированной патологией (недостаточный выброс левого желудочка) привело к значимому росту экскреции натрия с мочой (кратковременный эффект). Также повысилась концентрация в моче цикличного гуанозинмонофосфата, снизилась концентрация пептидов в плазме.
Использование комплексного средства в течение трех недель дало следующие результаты:
- повышение цикличного гуанозинмонофосфата;
- рост экскреции натрия;
- блокировку ангиотензивных рецепторов;
- уменьшение плазменной концентрации пептидов.
В исследовании сравнивались данные, полученные до и после лечения.
Полученный результат является показателем существенного увеличения интенсификации ренина и свидетельствует о росте его концентрации в плазме.
В сравнении с часто назначаемым Эналаприлом указанный комплекс – препарат Юперио приводил к более выраженному в целом терапевтическому результату.
Клиническая практика показала возможность для существенного уменьшения рисков летальных исходов при хронической сердечной недостаточности на фоне лечения комплексом. Отмечено уменьшение эпизодов лечения в стационаре страдающих коронарной недостаточностью (около 22% для группы, использующей комплекс и почти 27% для группы, принимающей эналаприл). Что касается рисков смертности из-за осложнений основного заболевания или регулярно повторяющихся эпизодов лечения в стационаре были получены следующие цифры – около 5% (из них: чуть более 3% — смерти из-за патологий сердца и сосудов и около 3% — пациенты с коронарной недостаточностью, госпитализированные впервые).
Уменьшение рисков (сравнение с эналаприлом): составляет 20 процентов.
При этом эффективность наблюдается уже в первые дни терапии и обнаруживает тенденцию к стабильному сохранению в течение всего срока лечения (убедительные данные получены в ходе клинических испытаний).
Эффект стал возможен благодаря синергии двух активных компонентов, взаимно усиливающих действие друг друга.
Эпизоды внезапного смертельного исхода (ранее были зарегистрированы данные: 45 процентов от числа всех случаев, вызванных сердечно-сосудистыми патологиями) уменьшились на 20 процентов (для группы пациентов, принимающих комплекс по сравнению с группой, использующей эналаприл).
Комплексный препарат (сакубитрил+валсартан) имеет высокую биодоступность в сравнении с другими таблетками с валсартаном в составе. Связывание с белками плазмы – до 97%.
Прием пищи не влияет на терапевтический эффект.
Попадая в организм сакубитрил преобразуется в сакубитрилат под действем ферментов. Далее существенного метаболизма не происходит. Валсартан незначительно метаболизируется (не более 20% от однократной дозы регистрируется в виде метаболитов).
Выведение
Почками: 13% (валсартан/метаболиты);
С калом: около 48% (сакубитрил), 86% (валсартан/метаболиты).
Показания к применению
Комплекс гипотензивный и кардиопротективный таблетки Юперио назначают при хронической сердечной недостаточности. Лекарство предназначено для уменьшения возможных рисков летального исхода, вызванных сердечно-сосудистыми патологиями, а также снижения частоты госпитализаций, связанных с диагнозом «коронарная недостаточность». При этом максимальный эффект зафиксирован у больных с недостаточным выбросом левого желудочка.
Показано применение комплекса также при эссенциальной артериальной гипертензии.
Противопоказания
Не назначаются таблетки в случае:
- гиперчувствительности к действующим компонентам и/или любым иным из вспомогательных веществ (см. список состава);
- приема других лекарственных препаратов, предназначенных для стабилизации повышенного артериального давления при стойкой артериальной гипертензии или терапии коронарной недостаточности. Рекомендован прием комплекса как минимум по истечению 1,5 суток после приема завершающей дозы таблеток, относящихся к указанной гр.;
- эпизода отека Квинке в прошлом и ряда генетических патологий, при которых ранее наблюдался отек Квинке;
- второго типа сахарного диабета, особенно при условии сопутствующего осложнения (умеренные или тяжелые функциональные почечные нарушения), а также при условии лечения препаратами с алискиреном в составе;
- тяжелого нарушения печеночной функции, включая билиарные циррозы (форма хронического аутоиммунного заболевания, возникающего вследствие сбоя процесса оттока желчи, а также нарушения ее выделения);
- возраста пациента до 18 лет;
- беременности или планирования зачатия, во время грудного вскармливания;
- одновременного применения любых иных препаратов с АРА II в составе.
При назначении комплекса следует учитывать вышеперечисленную информацию и исходить из имеющихся данных. В случае появления любых нежелательных реакций необходимо уведомить о проблемах лечащего врача.
Побочные действия
Во время приема комбинированного медикамента возможно появление нежелательных реакций.
Негативные эффекты
| Реакции, связанные с корректировкой доз или прекращением лечения | |
| Сосудистые реакции | Артериальная гипотензия Гипокалиемия Функциональные почечные нарушения ГиперкалиемияОбмороки Гипотония |
| ЖКТ | Тошнота Понос |
| Органы дыхания | Кашель |
| Почки | Острая почечная недостаточность Функциональные нарушения |
| Иммунная система | Гиперреакции Высыпания на коже, зуд Анафилаксия |
| Нервная система | Головокружение Головные боли |
| Прочее | Повышенная утомляемость Слабость Лабиринтные нарушения |
Юперио, инструкция по применению (Способ и дозировка)
Комплекс – 50 мг и 100 мг может быть использован только по назначению специалиста и только согласно правилам приема.
Препарат принимают внутрь – ежедневно, желательно в одно и то же время.
Таблетку нельзя делить на части, растирать, ломать и т.д.
Запивать водой, проглатывая целиком.
| Задача | Дозировки и курс |
| Лечение коронарной недостаточности | Юперио 200 мг дважды в день (одна табл. утро/вечер). Начальный курс: Юперио 100 мг дважды в сут. |
| Стабилизация показателей АД при артериальной гипертензии | 200 мг (однократно, 1т./сут,) При необходимости дозировки увеличиваются до 400 миллиграмм (однократно) |
Общие правила применения
Лечение проводится в виде монотерапии или в комплексе с иными гипотензивными таблетками.
Специалист подбирает адекватную дозу на основе имеющегося диагноза, состояния здоровья на момент обращения, показателей АД и т.д. Учитывается прием других препаратов пациентом, страдающим от любых сопутствующих патологий, а также взаимодействие лекарств.
Коррекции доз для пожилых пациентов не требуется. После определения наиболее подходящей начальной дозы для всех возрастных групп во многих случаях может понадобиться ее коррекция, учитывая индивидуальный ответ пациента на комплекс. Таким образом подбирается наиболее подходящий вариант дозировок. Обычно лечение начинают с Юперио 50 мг. Пациенты с сердечно-сосудистыми патологиями со стабильно высоким артериальным давлениям обязаны поддерживать постоянный контакт с лечащим врачом и информировать специалиста о своем состоянии.
Продолжительность лечения
Принимать комплекс следует постоянно (до отмены лечащим врачом).
Передозировка
Проверенной информации в медицинских источниках о случаях передозировки недостаточно для формирования оперативного мнения о негативных последствиях.
Однократная доза – 1200 мг и многократные дозы – 900 мг на протяжении двух недель у группы добровольцев с нормальными показателями состояния здоровья не стали причиной для какого-либо беспокойства по поводу негативного воздействия активных компонентов препарата на организм.
По мнению специалистов, вероятной симптоматикой передозировки в ряде случаев следует считать артериальную гипотензию, так как средство проявляет антигипертензивное действие.
К действенным лечебным мерам необходимо отнести симптоматическую терапию, проводимую под контролем врача. Выводить действующие вещества с помощью гемодиализа нецелесообразно по причине их связывания с белками плазмы крови.
Взаимодействие
Крайне нежелательно комбинировать средство с лекарственными препаратами, указанными ниже.
- Ингибиторы ангиотензинпревращающего фермента, к которым относятся такие широко применяющиеся препараты как Каптоприл, Эналаприл и их аналоги. Причина – возможен ангионевротический отек. Прием препарата разрешен по истечению 36 часов после прекращения терапии при помощи иАПФ. Такую же временную конфигурацию необходимо применять и в обратном порядке.
- Алискиренсодержащие препараты при наличии у пациента сахарного диабета или нарушенной функциональности почек.
Ряд лекарственных средств, не рекомендуемых для комбинированной терапии:
- Блокаторы ангиотензиновых рецепторов.
- Одновременное применение двух препаратов, содержащих АРА II считается нецелесообразным ввиду возможных побочных эффектов.
Лекарственные средства, которые следует использовать в комплексном лечении с осторожностью:
- Гиполипидемические препараты. Один из компонентов – сакубитрил подавляет действие переносчиков OATP1B1 и OATP1B3. Практика показала, что препарат увеличивает уровень статинов. При применении комбинированной терапии вместе с Симвастатином негативной динамики не зафиксировано.
- Силденафил. Комбинирование с силденафилом часто приводит к усилению антигипертензивного действия. Применять ингибиторы ФДЭ5 необходимо с учетом этого фактора.
Предположительная форма лекарственного взаимодействия с нижеследующими препаратами:
- Триамтерен и Амилорид, Спиронолактон и Эплеренон, препараты, содержащие калий, заменители соли. Перечисленные средства повышают уровень калия и креатинина. При необходимости применения комплексного лечения обязателен контроль содержания калия в сыворотке крови.
- Нестероиды. Применять их в комплексном лечении необходимо при учете возрастных ограничений (люди пенсионного возраста), возможной гиповолемии (применение диуретиков), нарушений функциональности почек – ввиду возможного нанесения вреда. Лечение – только под строгим контролем специалиста.
- Литий. Препараты, содержащие данный металл и ингибиторы АПФ и АРА II при одновременном применении создают фактор обратимого повышения количества лития в сыворотке крови. Возможный результат – токсические проявления, нередко достаточно тяжелые. Во время лечения необходим контроль количества металла в сыворотке крови – диуретические лекарственные препараты усиливают его токсическое действие.
- Рифампицин и Циклоспорин, а также Ритонавир. Препараты усиливают системную экспозицию сакубитрила/валсартана. Проявлять особую осторожность на начальной и завершающей стадии комплексного лечения.
В случае проявления негативной симптоматики при применении комплекса на фоне приема других лекарственных средств необходимо проконсультироваться с лечащим врачом и узкими специалистами.
Условия продажи
Рецепт от врача необходим.
Условия хранения
Температурный режим – от 10°C до 25 °C.
Обеспечить надежное хранение в темном месте. Оберегать от детей и подростков. Не применять по истечению срока годности. Утилизировать только в специально предназначенном месте. Берегите окружающую среду!
Срок годности
Предписано использовать таблетки на протяжении трех лет от даты выпуска. Не храните медикамент после истечения срока годности.
Особые указания
Пациентам с выраженным снижением артериального давления комплекс следует принимать с особой осторожностью. В медицинской практике отмечена клинически выраженная артериальная гипотензия у целого ряда пациентов. Снижение дозы диуретика, как дополнения к гипотензивному средству, в таких случаях быстро устраняет проблему. Более радикальная мера – уменьшение дозы или временный отказ от таблеток.
Контроль приема требуется в первую очередь пациентам, страдающим от гиповолемии. Им следует снизить на время прием диуретиков и отказаться от диеты с малым потреблением соли, обратить внимание на возможные рвотные позывы и симптомы диареи, если таковые появляются. Перед комплексным лечением необходимо проконтролировать уровень натрия в организме.
Функциональность почек на фоне терапии может ухудшаться, как правило, в незначительной степени. Случаи отмены лечения по этой причине довольно редки. Уменьшение дозы снимает проблематику вполне успешно. Пациенты со значительными нарушениями функций почек должны принимать гипотензивное средство под контролем лечащего врача.
Развитие гиперкалиемии наблюдается в редких случаях. С осторожностью следует назначать средства, увеличивающие содержание калия. Рациональным решением считается употребление продуктов с небольшим количеством калия и уменьшение дозы медикаментов, входящих в комплексную терапию.
Контроль содержания калия в сыворотке крови обязателен при употреблении продуктов с высоким содержанием калия, а также при наличии тяжелых нарушений функций почек, сахарного диабета, гипоальдостеронизма.
Симптоматика ангионевротического отека – серьезная причина для отмены лечения и отказ от повторного прием таблеток. Лечение – симптоматическое. Особое внимание следует обратить на возможный отек гортани, языка, губ. Исключить обструкцию можно введением раствора эпинефрина: 1:1000 (0.3-0.5 мл) и обеспечением надлежащих комплексных мер по экстренной помощи. Перед назначением таблеток следует выяснить, нет ли у пациента наследственной предрасположенности к ангионевротическим отекам. В таких случаях применение средства запрещено.
При наличии стеноза почечной артерии прием комплекса должен осуществляться вместе с контролем функций почек.
Управление транспортным средством и/или механизмом разрешено совмещать с активным лечением, так как негативного влияния на концентрацию внимания и координацию движений в медицинской практике не зафиксировано. Следует соблюдать осторожность при наличии симптомов, указывающих на переутомление и повышенное АД и т.д.
Аналоги Юперио
Совпадения по коду АТХ 4-го уровня:
В аптеках РФ можно приобрести 93 сходных по действию медикаментов – 2 групповых и 90 нозологических.
Полные аналоги отсутствуют.
Групповые аналоги: Ко-Вамлосет, Ко-Эксфорж.
Перечисленные аналоги предлагаются в форме таблеток для внутреннего применения.
Для детей
Прием препарата разрешен по достижению возраста 18 лет.
С алкоголем
Употребления алкогольных напитков противопоказано.
При беременности и лактации
Половые контакты на фоне лечения и на протяжении семи дней после терапии допускаются с применением надежного средства контрацепции.
Во время вынашивания плода от таблеток следует отказаться. Причина – возможное прерывание беременности, маловодие, функциональные нарушения почек у новорожденного.
Матерям, кормящим грудью, лечение с помощью указанного комплекса в большинстве случаев противопоказано.
Снижение фертильности не зафиксировано.
Отзывы об Юперио
Основная причина покупки таблеток, по мнению обычных людей и специалистов – поиск наиболее эффективного индивидуального средства для стабилизации артериального давления.
Отзывы кардиологов в целом положительные. Возникшие побочные эффекты, как правило, результат неграмотного выбора, самолечения, а также несоблюдения врачебных предписаний и указаний в инструкции.
Отзывы пациентов основываются на личном опыте, субъективных предположениях и ощущениях. Выбор оптимального лекарственного средства на их основании требует, как минимум, профессиональной коррекции. Это касается в первую очередь людей, страдающих от хронической сердечной недостаточности.
Цена Юперио, где купить
Стоимость лекарственного средства в аптечных сетях Москвы варьируется:
- от 2300 до 3000 рублей за пачку 200 мг (28 таблеток);
- от 4400 до 5000 рублей за пачку 200 мг (56 таблеток).
Стоимость комплексного препарата и других гипотензивных и кардиопротективных средств в столице и регионах РФ может сильно отличаться.
- Интернет-аптеки РоссииРоссия
ЗдравСити
-
Юперио таблетки п/о плён. (25,7мг+24,3мг) 50мг 28штООО Новартис Нева
-
Юперио таблетки п/о плён. (51,4мг+48,6мг) 100мг 28штООО Новартис Нева
-
Юперио таблетки п/о плён. (102,8мг+97,2мг) 200мг 28штООО Новартис Нева
-
Юперио таблетки п/о плён. (25,7мг+24,3мг) 50мг 56штООО Новартис Нева
-
Юперио таблетки п/о плён. (102.8мг+97.2мг) 200мг 56штООО Новартис Нева
Аптека Диалог
-
Юперио таблетки 100мг №56Новартис Нева ООО
-
Юперио таблетки 200 мг (102.8 мг+97.2 мг) №56Новартис Нева ООО
-
Юперио таблетки 100мг №28Новартис Нева ООО
-
Юперио таблетки 50мг №28Novartis Farma/Новартис Нева ООО
-
Юперио таблетки 200мг №28Novartis Farma
показать еще
До начала терапии препаратом Пралуэнт необходимо исключить вторичные причины гиперлипидемии или смешанной дислипидемии (например, нефротический синдром, гипотиреоз).
В период лечения препаратом Пралуэнт пациент должен продолжать соблюдать диету, снижающую уровень холестерина.
Препарат вводят п/к в переднюю часть бедра, плеча или переднюю часть живота.
Начальная доза препарата Пралуэнт составляет 75 мг 1 раз каждые 2 недели. У пациентов, которым требуется большее снижение концентрации ХС-ЛПНП (> 60%), начальная доза препарата Пралуэнт может составлять 150 мг, которую также вводят 1 раз каждые 2 недели или 300 мг 1 раз каждые 4 недели (ежемесячно).
Дозу препарата Пралуэнт следует подбирать индивидуально на основании таких параметров как исходные значения ХС-ЛПНП, цели терапии и ответ пациента на лечение. Концентрации липидов в крови можно оценивать через 4-8 недель после начала лечения или титрования дозы и проводить соответствующую коррекцию дозы. При необходимости дополнительного снижения концентрации ХС-ЛПНП у пациентов, которым препарат Пралуэнт назначался в дозе 75 мг 1 раз каждые 2 недели или 300 мг 1 раз каждые 4 недели, доза может быть скорректирована до максимальной дозы 150 мг 1 раз каждые 2 недели.
В случае пропуска очередного введения препарата, пациенту необходимо как можно скорее ввести пропущенную дозу и затем продолжить терапию в соответствии с исходным режимом дозирования.
Пациенты, которым проводится ЛПНП-аферез
Препарат Пралуэнт может применяться у пациентов, которым проводится ЛПНП-аферез, независимо от времени и частоты проведения процедуры. Рекомендуемая доза — 150 мг 1 раз каждые 2 недели. Концентрации ХС-ЛПНП можно оценивать до афереза для определения необходимости его проведения.
Особые группы пациентов
Пациенты пожилого возраста. У пациентов пожилого возраста коррекции дозы препарата Пралуэнт не требуется (см. раздел «Особые указания»).
Печеночная недостаточность. У пациентов с печеночной недостаточностью легкой или средней степени тяжести коррекции дозы препарата Пралуэнт не требуется. Данные о применении препарата Пралуэнт у пациентов с печеночной недостаточностью тяжелой степени отсутствуют.
Почечная недостаточность. У пациентов с почечной недостаточностью легкой или средней степени тяжести коррекции дозы препарата Пралуэнт не требуется. Данные о применении препарата у пациентов с почечной недостаточностью тяжелой степени ограничены.
Масса тела. Не требуется коррекции режима дозирования в зависимости от массы тела пациентов.
Правила введения препарата
Препарат Пралуэнт применяют в виде п/к инъекций, проводимых в области бедра, живота или плеча, с помощью одноразовой предварительно заполненной шприц-ручки или одноразового предварительно заполненного шприца.
Рекомендуется менять места инъекций при проведении каждой инъекции.
При назначении дозы 300 мг препарат вводят в виде двух инъекций по 150 мг в разные места.
Препарат Пралуэнт не следует вводить в области активных кожных заболеваний или повреждений, таких как солнечные ожоги, кожная сыпь, воспаления кожи или кожные инфекции.
Не следует вводить препарат Пралуэнт в то же место, в которое вводились другие лекарственные препараты.
Правила обращения с препаратом
Хранить при температуре от 2°С до 8°С. Не замораживать.
Не подвергать воздействию высоких температур. При необходимости (например, во время путешествия) препарат Пралуэнт можно хранить при температуре не выше 25°С не более 30 дней. Защищать от света. После извлечения из холодильника препарат Пралуэнт должен быть использован в течение 30 дней или утилизирован.
Описание предварительно заполненных одноразовых шприц-ручек и инструкция по их использованию
Пралуэнт, раствор для п/к введения 75 мг/мл в предварительно заполненной шприц-ручке по 1 мл
Пралуэнт, раствор для п/к введения 150 мг/мл в предварительно заполненной шприц-ручке по 1 мл
Инструкция по применению
Пациент должен быть информирован о том, что он должен сохранять эту инструкцию, т.к. она может понадобиться ему снова. Если у пациента появятся вопросы, то ему следует обратиться к лечащему врачу или провизору, или позвонить по номеру телефона, указанному в листке-вкладыше.
Части шприц-ручки показаны на рисунке:
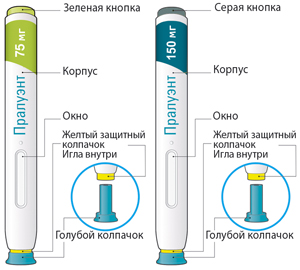
Важная информация
Шприц-ручка предназначена только для одноразового использования.
Шприц-ручка с зеленой кнопкой: в 1 мл раствора содержится 75 мг алирокумаба.
Шприц-ручка с серой кнопкой: в 1 мл раствора содержится 150 мг алирокумаба.
Препарат вводится п/к и может вводиться пациентом самостоятельно или другим лицом, ухаживающим за ним.
Шприц-ручку можно использовать только для одноразовой инъекции, и она должна быть утилизирована после применения.
Правила использования
Шприц-ручку следует хранить в недоступном для детей месте.
Перед использованием шприц-ручки следует внимательно прочитать инструкцию.
Необходимо следовать всем указаниям, представленным в инструкции, при каждом использовании шприц-ручки.
Неиспользованные шприц-ручки следует хранить в холодильнике при температуре от 2°С до 8°С.
Запрещено
Не трогайте желтый защитный колпачок.
Не используйте шприц-ручку при протекании или повреждении.
Не используйте шприц-ручку при отсутствии голубого колпачка, или если он ненадежно закреплен.
Не используйте шприц-ручку повторно.
Не трясите шприц-ручку.
Не замораживайте шприц-ручку.
Не подвергайте шприц-ручку воздействию сильного тепла и прямых солнечных лучей.
ЭТАП А: ПОДГОТОВКА К ВВЕДЕНИЮ
Перед введением препарата понадобятся:
- шприц-ручка с препаратом Пралуэнт;
- салфетки, смоченные спиртом;
- ватные тампоны или марля;
- контейнер, устойчивый к проколам.
1. Проверьте этикетку на шприц-ручке
Проверьте, что Вы взяли правильный (нужный Вам) препарат и правильную (нужную Вам) дозу (шприц-ручка с зеленой кнопкой для дозы 75 мг/мл и шприц-ручка с серой кнопкой для дозы 150 мг/мл).
Проверьте срок годности. Не использовать по истечении срока годности.
2. Проверьте окно
Проверьте, чтобы видимая в окно жидкость была прозрачной, бесцветной или бледно-желтой, без включений. Если имеются изменения цвета и видимые включения, то не следует использовать шприц-ручку (см. рисунок А).
Вы можете увидеть пузырьки воздуха. Это нормальное явление и не влияет на дальнейшее использование препарата.
Не используйте шприц-ручку, если окно желтого цвета и не содержит жидкости (см. рисунок В).
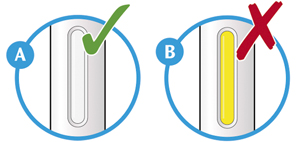
3. Оставьте шприц-ручку нагреваться при комнатной температуре в течение 30-40 мин
Не нагревайте шприц-ручку, она должна нагреться сама.
Используйте шприц-ручку как можно скорее после того, как она нагрелась.
Не кладите шприц-ручку обратно в холодильник
4. Подготовка места введения препарата
Вымойте руки с мылом и водой и вытрите их полотенцем.
Вы можете вводить препарат (см. рисунок):
- в область бедра;
- в область живота (за исключением области 5 см вокруг пупка);
- в наружную поверхность плеча.
Вы можете стоять или сидеть во время введения препарата.
Продезинфицируйте место введения салфеткой, смоченной спиртом.
Не вводите препарат в болезненную, уплотненную, покрасневшую или воспаленную кожу.
Не вводите препарат в места рядом с видимыми венами.
Используйте разные места при каждом введении препарата.
Не вводите препарат в одно и то же место с другими препаратами.
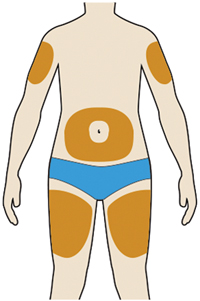
ЭТАП В: КАК ВВОДИТЬ ПРЕПАРАТ
1. После завершения всех пунктов «ЭТАП А: ПОДГОТОВКА К ВВЕДЕНИЮ», снимите голубой колпачок.
Не снимайте колпачок до тех пор, пока Вы не будете готовы к введению препарата.
Не надевайте голубой колпачок обратно.
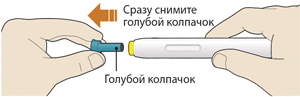
2. Держите шприц-ручку, как показано на рисунке.
Не трогайте желтый защитный колпачок.
Убедитесь, что Вы видите окно.
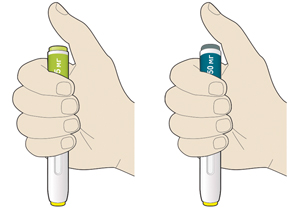
3. Прижмите желтый защитный колпачок к коже примерно под углом 90°
Прижимайте шприц-ручку к коже и удерживайте ее так, чтобы желтый защитный колпачок стал невидимым (вдавленным в кожу). Шприц-ручка не будет работать, если желтый защитный колпачок не вдавлен в кожу полностью.
При необходимости, зажмите кожу, чтобы убедиться, что место введения плотное.
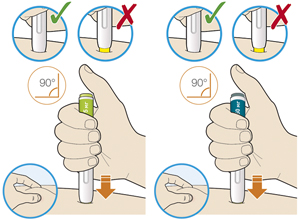
4. Нажмите большим пальцем на зеленую кнопку, если Вы используете шприц-ручку Пралуэнт 75 мг, или на серую кнопку, если Вы используете шприц-ручку Пралуэнт 150 мг, и сразу отпустите ее.
Вы услышите щелчок. Введение препарата началось.
Окно начнет желтеть.
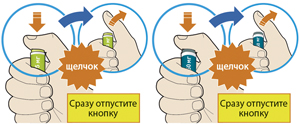
5. Продолжайте удерживать шприц-ручку, плотно прижатой к коже после того, как отпустили кнопку
Введение препарата может продолжаться до 20 сек.
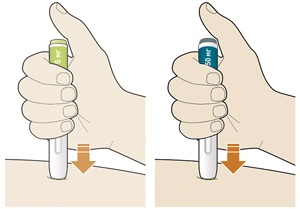
6. Проверьте, что окно стало желтого цвета до того, как уберете шприц-ручку от кожи
Не убирайте шприц-ручку от кожи до тех пор, пока все окно не будет желтого цвета.
Введение препарата завершено после того, как все окно будет желтого цвета, и Вы можете услышать второй щелчок.
Если окно не стало полностью желтого цвета, извлеките шприц-ручку и обратитесь за помощью к лечащему врачу.
Пациент не должен делать себе вторую инъекцию, не посоветовавшись с лечащим врачом.
7. Уберите шприц-ручку от кожи
Не растирайте кожу после введения препарата.
Если Вы видите кровь, прижмите ватный тампон или марлю и удерживайте их до тех пор, пока не остановится кровотечение.
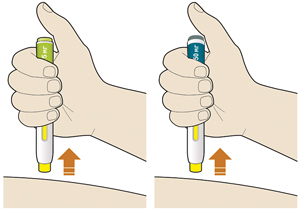
8. Утилизируйте шприц-ручку и колпачок
Не надевайте голубой колпачок повторно.
Поместите шприц-ручку в контейнер, устойчивый к проколам.
Спросите у лечащего врача, как утилизировать контейнер.
Всегда храните контейнер в недоступном для детей месте.
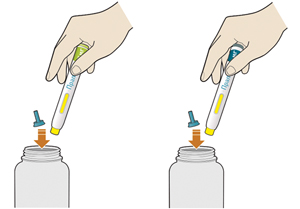
ПОДГОТОВКА И ОБРАЩЕНИЕ С ПРЕПАРАТОМ
Пациент может вводить препарат Пралуэнт самостоятельно, или препарат может быть введен лицом, ухаживающим за пациентом, после предоставления им медицинскими работниками информации по правильной технике проведения п/к инъекций.
Препараты для парентерального введения должны осматриваться визуально перед введением на предмет наличия видимых частиц и изменения цвета. Если раствор имеет измененный цвет или видимые частицы, его не следует использовать.
Перед использованием предварительно заполненных шприц-ручек следует нагреть их до комнатной температуры. При необходимости (например, во время путешествия) препарат Пралуэнт можно хранить при температуре не выше 25°С не более 30 дней. Защищать от света. После извлечения из холодильника препарат Пралуэнт должен быть использован в течение 30 дней или утилизирован. После использования шприц-ручки следует поместить в устойчивый к проколам контейнер и утилизировать согласно местным (государственным) требованиям. Не использовать контейнер повторно. Всегда следует хранить контейнер в местах, недоступных для детей.
Пациенты и ухаживающие за ними лица должны получить руководство по надлежащей технике п/к введения, включая сведения по асептике, и по тому, как правильно использовать предварительно заполненные шприц-ручки.
Пациентов и ухаживающих за ними лиц следует информировать о том, что перед первым введением препарата Пралуэнт они должны внимательно прочитать инструкцию по применению препарата.
Пациентов и ухаживающих за ними лиц следует предупредить, что запрещено повторное использование шприц-ручек, и они должны быть проинструктированы, как безопасно утилизировать их после использования.
Пралуэнт, раствор для п/к введения 150 мг/мл в предварительно заполненной шприц-ручке по 2 мл
Инструкция по применению
Пациент должен быть информирован о том, что он должен сохранять эту инструкцию, т.к. она может понадобиться ему снова. Если у пациента появятся вопросы, то ему следует обратиться к лечащему врачу или провизору, или позвонить по номеру телефона, указанному в листке-вкладыше.
Части шприц-ручки показаны на рисунке:
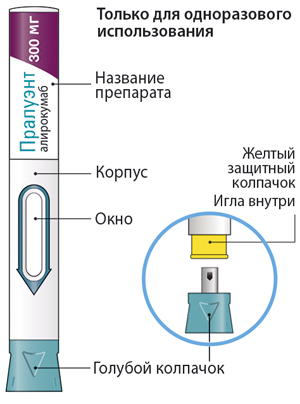
Важная информация
Шприц-ручка предназначена только для одноразового использования; в 2 мл раствора содержится 300 мг алирокумаба.
Препарат вводится п/к и может вводиться пациентом самостоятельно или другим лицом, ухаживающим за ним.
Перед использованием шприц-ручки пациент должен пройти обучение у лечащего врача.
Шприц-ручку можно использовать только для одноразовой инъекции, и она должна быть утилизирована после применения.
Правила использования
Шприц-ручку следует хранить в недоступном для детей и защищенном от света месте.
Перед использованием шприц-ручки следует внимательно прочитать инструкцию.
Необходимо следовать всем указаниям, представленным в инструкции, при каждом использовании шприц-ручки.
Неиспользованные шприц-ручки следует хранить в холодильнике при температуре от 2°С до 8°С.
Запрещено
Не трогайте желтый защитный колпачок.
Не используйте шприц-ручку, если она протекает или повреждена.
Не используйте шприц-ручку при отсутствии голубого колпачка, или если он ненадежно закреплен.
Не используйте шприц-ручку повторно.
Не трясите шприц-ручку.
Не замораживайте шприц-ручку.
Не подвергайте шприц-ручку воздействию тепла или прямых солнечных лучей.
ЭТАП А: ПОДГОТОВКА К ВВЕДЕНИЮ
Перед введением препарата понадобятся:
- шприц-ручка с препаратом Пралуэнт;
- салфетки, смоченные спиртом;
- ватные тампоны или марля;
- контейнер, устойчивый к проколам.
1. Проверьте этикетку на шприц-ручке
Проверьте, что Вы взяли правильный (нужный Вам) препарат и правильную (нужную Вам) дозу.
Проверьте срок годности. Не использовать по истечении срока годности.

2. Проверьте окно
Проверьте, чтобы видимая в окно жидкость была прозрачной, бесцветной или бледно-желтой, без включений. Если имеются изменения цвета и видимые включения, то не следует использовать шприц-ручку (см. рисунок А).
Вы можете увидеть пузырьки воздуха. Это нормальное явление и не влияет на дальнейшее использование препарата.
Не используйте шприц-ручку, если окно желтого цвета и не содержит жидкости (см. рисунок В).
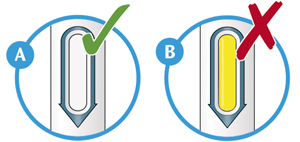
3. Оставьте шприц-ручку нагреваться при комнатной температуре в течение 45 мин.
Это важно для применения всей дозы и помогает минимизировать дискомфорт при введении препарата.
Не нагревайте шприц-ручку, она должна нагреться сама.
Не кладите шприц-ручку обратно в холодильник.
4. Подготовка места введения препарата
Вымойте руки с мылом и водой и вытрите их полотенцем.
Вы можете вводить препарат (см. рисунок):
- в верхнюю часть бедра;
- в область живота (за исключением области 5 см вокруг пупка);
- в наружную поверхность плеча (может быть сделано только лицом, ухаживающим за Вами).
Вы можете стоять или сидеть во время введения препарата.
Продезинфицируйте место введения салфеткой, смоченной спиртом.
Не вводите препарат в болезненную, уплотненную, покрасневшую или воспаленную кожу.
Не вводите препарат в места рядом с видимыми венами.
Изменяйте места инъекций при каждом введении препарата (в т.ч. в области введения).
Не вводите препарат в одно и то же место с другими препаратами.
ЭТАП В: КАК ВВОДИТЬ ПРЕПАРАТ
1. После завершения всех пунктов «ЭТАП А: ПОДГОТОВКА К ВВЕДЕНИЮ», снимите голубой колпачок.
Не снимайте голубой колпачок до тех пор, пока Вы не будете готовы к введению препарата.
Не надевайте голубой колпачок обратно.
Не используйте шприц-ручку при отсутствии голубого колпачка, или если он ненадежно закреплен.
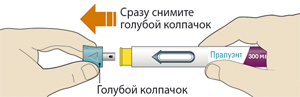
2. Держите шприц-ручку, как показано на рисунке.
Не трогайте желтый защитный колпачок. Игла находится внутри желтого защитного колпачка.
Убедитесь, что Вы видите окно.
Не прижимайте шприц-ручку к коже, пока не будете готовы к введению препарата.
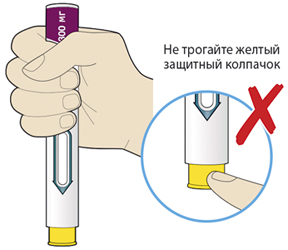
3. Прижмите желтый защитный колпачок к коже примерно под углом 90°
Зажмите кожу, чтобы убедиться, что место введения плотное.
Прижимайте шприц-ручку к коже до тех пор, пока желтый защитный колпачок не войдет в шприц-ручку до упора, и удерживайте ее (см. рисунок).
Введение препарата не начнется, пока желтый защитный колпачок не будет полностью вжат.
Вы услышите щелчок. Введение препарата началось. Окно начнет желтеть.
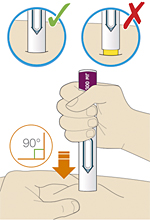
4. Продолжайте удерживать шприц-ручку, плотно прижатой к коже
Вы можете услышать второй щелчок.
Убедитесь, что окно стало желтым.
Затем медленно посчитайте до 5.
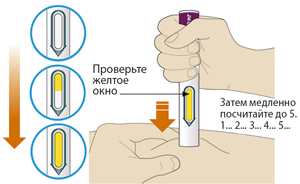
5. Проверьте, что окно стало желтого цвета до того, как уберете шприц-ручку от кожи
Если окно не стало полностью желтым, извлеките шприц-ручку и обратитесь за помощью к лечащему врачу.
Пациент не должен делать себе вторую инъекцию, не посоветовавшись с лечащим врачом.
6. Уберите шприц-ручку от кожи
Не растирайте кожу после введения препарата.
Если Вы видите кровь, прижмите ватный тампон или марлю и удерживайте их до тех пор, пока не остановится кровотечение.
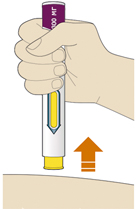
7. Утилизируйте шприц-ручку и колпачок
Не надевайте голубой колпачок повторно.
Сразу же после введения препарата поместите шприц-ручку в контейнер, устойчивый к проколам.
Спросите у лечащего врача, как утилизировать контейнер.
Всегда храните контейнер в недоступном для детей месте.
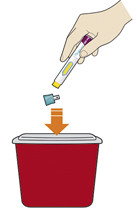
ПОДГОТОВКА И ОБРАЩЕНИЕ С ПРЕПАРАТОМ
Пациент может вводить препарат Пралуэнт самостоятельно, или препарат может быть введен лицом, ухаживающим за пациентом, после предоставления им медицинскими работниками информации по правильной технике проведения п/к инъекций.
Препараты для парентерального введения должны осматриваться визуально перед введением на предмет наличия видимых частиц и изменения цвета. Если раствор имеет измененный цвет или видимые частицы, его не следует использовать.
Перед использованием предварительно заполненной шприц-ручки следует нагреть ее до комнатной температуры. При необходимости (например, во время путешествия) препарат Пралуэнт можно хранить при температуре не выше 25°С не более 30 дней. Защищать от света. После извлечения из холодильника препарат Пралуэнт должен быть использован в течение 30 дней или утилизирован. После использования шприц-ручки следует поместить в устойчивый к проколам контейнер и утилизировать согласно местным (государственным) требованиям. Не использовать контейнер повторно. Всегда следует хранить контейнер в местах, недоступных для детей.
Пациенты и ухаживающие за ними лица должны получить руководство по надлежащей технике п/к введения, включая сведения по асептике, и по тому, как правильно использовать предварительно заполненные шприц-ручки.
Пациентов и ухаживающих за ними лиц следует информировать о том, что перед первым введением препарата Пралуэнт они должны внимательно прочитать инструкцию по применению препарата.
Пациентов и ухаживающих за ними лиц следует предупредить, что запрещено повторное использование шприц-ручек, и они должны быть проинструктированы, как безопасно утилизировать их после использования.
Описание предварительно заполненных одноразовых шприцев и инструкция по их использованию
Пралуэнт, раствор для п/к введения 75 мг/мл в предварительно заполненном шприце
Пралуэнт, раствор для п/к введения 150 мг/мл в предварительно заполненном шприце
Инструкция по применению
Пациент должен быть информирован о том, что он должен сохранять эту инструкцию, т.к. она может понадобиться ему снова. Если у пациента появятся вопросы, то ему следует обратиться к лечащему врачу или провизору, или позвонить по номеру телефона, указанному в листке-вкладыше.
Части шприца показаны на рисунке.
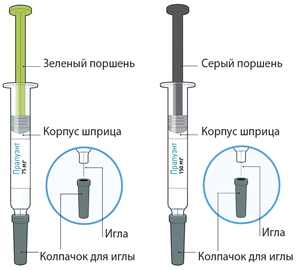
Важная информация
Шприц предназначен только для одноразового использования.
Шприц с зеленым поршнем: в 1 мл раствора содержится 75 мг алирокумаба.
Шприц с серым поршнем: в 1 мл раствора содержится 150 мг алирокумаба.
Препарат вводится п/к и может вводиться пациентом самостоятельно или другим лицом, ухаживающим за ним.
Шприц можно использовать только для одноразовой инъекции, и он должен быть утилизирован после применения.
Правила использования
Шприц следует хранить в недоступном для детей месте.
Перед использованием шприца следует внимательно прочитать инструкцию.
Необходимо следовать всем указаниям, представленным в инструкции, при каждом использовании шприца.
Неиспользованные шприцы следует хранить в холодильнике при температуре от 2°С до 8°С.
Запрещено
Не трогайте иглу.
Не используйте шприц при протекании или повреждении.
Не используйте шприц при отсутствии серого колпачка для иглы, или если он ненадежно закреплен.
Не используйте шприц повторно.
Не трясите шприц.
Не замораживайте шприц.
Не подвергайте шприц воздействию прямых солнечных лучей.
ЭТАП А: ПОДГОТОВКА К ВВЕДЕНИЮ
Перед началом введения понадобятся:
- шприц с препаратом Пралуэнт;
- салфетки, смоченные спиртом;
- ватные тампоны или марля;
- контейнер, устойчивый к проколам.
1. Перед началом
Возьмите шприц, удерживая его за корпус.
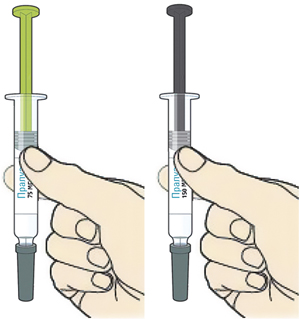
2. Проверьте этикетку на шприце
Проверьте, что Вы взяли правильный (нужный Вам) препарат и правильную (нужную Вам) дозу (шприц с зеленым поршнем для дозы 75 мг/мл и шприц с серым поршнем для дозы 150 мг/мл).
Проверьте срок годности: не использовать по истечении срока годности.
Проверьте, чтобы жидкость в шприце была прозрачной, бесцветной или бледно-желтой, без включений. Если имеются изменения цвета и видимые включения, то не следует использовать шприц.
Проверьте, что шприц не открыт и не поврежден.
3. Оставьте шприц нагреваться при комнатной температуре в течение 30-40 мин
Не нагревайте шприц, он должен нагреться сам.
Используйте шприц как можно скорее после того, как он нагреется.
Не кладите шприц обратно в холодильник.
4. Подготовка места введения
Вымойте руки с мылом и водой и вытрите их полотенцем.
Вы можете вводить препарат (см. рисунок):
- в область бедра;
- в область живота (за исключением области 5 см вокруг пупка);
- в наружную поверхность плеча.
Вы можете стоять или сидеть во время проведения инъекции.
Продезинфицируйте место введения салфеткой, смоченной спиртом.
Не вводите препарат в болезненную, уплотненную, покрасневшую или воспаленную кожу.
Не вводите препарат в места рядом с видимыми венами.
Используйте разные места при каждом введении препарата.
Не вводите препарат в одно и то же место с другими препаратами.
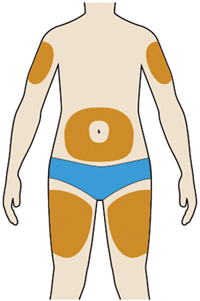
ЭТАП В: КАК ВВОДИТЬ ПРЕПАРАТ
1. После завершения всех пунктов «ЭТАП А: ПОДГОТОВКА К ВВЕДЕНИЮ» снимите колпачок для иглы
Не снимайте колпачок до тех пор, пока Вы не будете готовы к введению препарата.
Держите шприц за середину корпуса так, чтобы игла была направлена от Вас.
Не прикасайтесь к поршню.
Вы можете увидеть пузырьки воздуха. Это нормальное явление и не влияет на дальнейшее использование препарата. Не избавляйтесь ни от каких пузырьков в шприце до начала инъекции.
Не надевайте серый колпачок обратно.
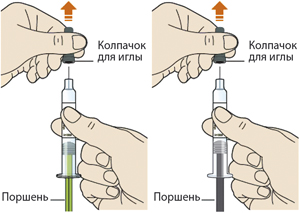
2. При необходимости зажмите кожу
Большим и указательным пальцем зажмите кожу так, чтобы получилась складка кожи в месте введения.
Удерживайте кожную складку во время проведения всей инъекции.
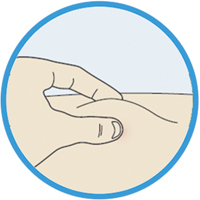
3. Введите иглу в складку кожу быстрым движением
Используйте угол в 90° при сжатии 5 см кожи.
Используйте угол в 45° при сжатии 2 см кожи.
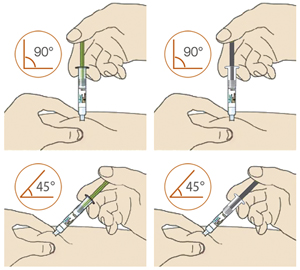
4. Опустите поршень вниз
Введите весь раствор медленным и равномерным нажатием на поршень.
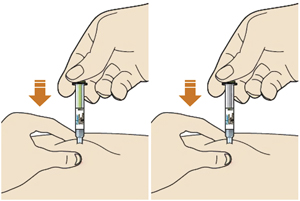
5. Перед тем как удалить иглу убедитесь, что шприц пуст
Не удаляйте шприц, пока он полностью не опустеет.
Извлекайте иглу из кожи под тем же углом, под каким она была введена.
Не растирайте кожу после введения препарата.
Если Вы видите кровь, прижмите ватный тампон или марлю и удерживайте их до тех пор, пока не остановится кровотечение.
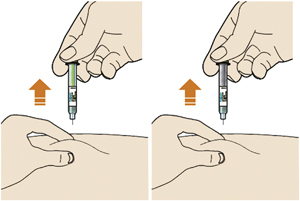
6. Утилизируйте шприц и колпачок.
Не надевайте серый колпачок повторно.
Не используйте шприц повторно.
Поместите шприц в контейнер, резистентный к проколам.
Всегда храните контейнер в недоступном для детей месте.
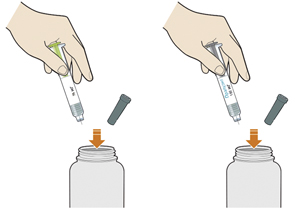
ПОДГОТОВКА И ОБРАЩЕНИЕ С ПРЕПАРАТОМ
Пациент может вводить препарат Пралуэнт самостоятельно, или препарат может быть введен лицом, ухаживающим за пациентом, после предоставления им медицинскими работниками информации по правильной технике проведения п/к инъекций.
Препараты для парентерального введения должны осматриваться визуально перед введением на предмет наличия видимых частиц и изменения цвета. Если раствор имеет измененный цвет или видимые частицы, его не следует использовать.
Перед использованием предварительно заполненных шприцев следует нагреть их до комнатной температуры. При необходимости (например, во время путешествия) препарат Пралуэнт можно хранить при температуре не выше 25°С не более 30 дней. Защищать от света. После извлечения из холодильника препарат Пралуэнт должен быть использован в течение 30 дней или утилизирован. После использования шприцы следует поместить в устойчивый к проколам контейнер и утилизировать согласно местным (государственным) требованиям. Не использовать контейнер повторно. Всегда следует хранить контейнер в местах, недоступных для детей.
Пациенты и ухаживающие за ними лица должны получить руководство по надлежащей технике п/к введения, включая сведения по асептике, и по тому, как правильно использовать предварительно заполненные шприцы.
Пациентов и ухаживающих за ними лиц следует информировать о том, что перед первым введением препарата Пралуэнт они должны внимательно прочитать инструкцию по применению препарата.
Пациентов и ухаживающих за ними лиц следует предупредить, что запрещено повторное использование шприцев, и они должны быть проинструктированы, как безопасно утилизировать их после использования.