
Внешний вид упаковки может отличаться от фотографии
Информация о товаре
Форма выпуска:
раствор для инфузий
Количество в упаковке:
1 шт.
Производитель:
Amgen Manufacturing
Страна:
Россия
Страна производства может отличаться, проверяйте при получении заказа
Популярные товары в категории
Популярные товары в Ютеке
Купить Этесевимаб, 700 мг/20 мл, раствор для инфузий, 1 шт. в Москве с доставкой в аптеку или домой, сделав заказ через Ютеку
Цена Этесевимаб, 700 мг/20 мл, раствор для инфузий, 1 шт. в Москве от 0 руб. на сайте и в приложении
Подробная инструкция по применению Этесевимаб, 700 мг/20 мл, раствор для инфузий, 1 шт.
Информация на сайте не является призывом или рекомендацией к самолечению и не заменяет консультацию специалиста (врача), которая обязательна перед назначением и/или применением любого лекарственного препарата.
Дистанционная торговля лекарственными препаратами осуществляется исключительно аптечными организациями, имеющими действующую лицензию на фармацевтическую деятельность, а также разрешение на дистанционную торговлю лекарственными препаратами. Дистанционная торговля рецептурными лекарственными препаратами, наркотическими и психотропными, а также спиртосодержащими лекарственными препаратами запрещена действующим законодательством РФ и не осуществляется.
Бамланивимаб + Этесевимаб
Bamlanivimab + Etesevimab
Фармакологическое действие
Бамланивимаб и этесевимаб связываются с разными, но перекрывающимися сайтами белка шипа вируса SARS-CoV-2.
Бамланивимаб — человеческое нейтрализующее моноклональное IgG1-антитело, нацеленное против S-белка SARS-CoV-2 (COVID-19). S-белок на поверхности вирусной частицы SARS-CoV-2 служит ключом для проникновения вируса в клетки хозяина. Являясь трансмембранным поверхностным гликопротеином S-белок помогает прикреплять вирион к мембране клетки-хозяина посредством взаимодействия с рецептором ангиотензинпревращающего фермента 2 (ACE2) на эпителиальных клетках дыхательных путей и клетках желудочно-кишечного тракта, что позволяет геному вируса проникнуть в клетку и запустить репликацию. Бамланивимаб связывает рецептор-связывающий домен (RBD) вирусного S-белка в эпитопе, частично перекрывающем сайт связывания ACE2, тем самым конкурируя с последним, что теоретически должно влиять на снижение продолжительности тяжести течения заболевания или сокращением продолжительности заболевания.
Этесевимаб — полностью человеческое рекомбинантное моноклональное антитело, нацелено на связывающий домен поверхностного шипового белка SARS-CoV-2 (COVID-19).
Показания
Для лечения лабораторно подтверждённой новой коронавирусной инфекции COVID-19 лёгкой и средней степени тяжести у взрослых и детей, включая новорождённых, и которые подвержены факторам высокого риска* развития тяжёлой формы COVID-19 и/или госпитализации.
* Факторы риска могут включать, но не ограничиваются:
- Пожилой возраст.
- Ожирение.
- Сердечно-сосудистые заболевания, включая артериальную гипертензию.
- Хронические заболевания лёгких, включая бронхиальную астму.
- Сахарный диабет 1 и 2 типов.
- Нарушение функции почек, включая пациентов, находящихся на диализе.
- Нарушение функции печени.
- Иммуносупрессивные состояния, основанные на оценке врача. Например, при лечении пациентов со злокачественными новообразованиями, после трансплантации костного мозга и солидных органов, при иммунодефиците, ВИЧ-инфекции (плохо контролируемая ВИЧ-инфекция или СПИД), серповидно- клеточной анемии, талассемии и пациентов, находящихся на длительном лечении иммуносупрессивными препаратами.
Способ применения и дозы
700 мг бамланивимаба (1 флакон) и 1400 мг этесевимаба (2 флакона) следует разбавлять и вводить вместе в виде однократной внутривенной инфузии.
Необходим контроль клинического состояния пациентов во время введения и наблюдение за ними в течение не менее 1 часа после завершения инфузии.
Побочные действия
Возможные побочные эффекты при одновременном применении бамланивимаба и этесевимаба включают тошноту, головокружение, зуд, сыпь.
Особые указания
Подробнее о лечении COVID-19
Временные методические рекомендации профилактики, диагностики и лечения новой коронавирусной инфекции (COVID-19)*
В список возможных к назначению лекарственных средств для лечения COVID-19 у взрослых включены:
- Фавипиравир,
- Молнупиравир,
- Нирматрелвир + Ритонавир,
- Ремдесивир,
- Синтетическая малая интерферирующая рибонуклеиновая кислота (миРНК) [двуцепочечная],
- Иммуноглобулин человека против COVID-19,
- Интерферон-альфа (IFN-α),
- Умифеновир,
- Имидазолилэтанамид пентандиовой кислоты,
- Касиривимаб + имдевимаб,
- Бамланивимаб + этесевимаб,
- Сотровимаб,
- Регданвимаб.
В список препаратов упреждающей противовоспалительной терапии COVID-19 у взрослых включены:
- Барицитиниб,
- Тофацитиниб,
- Упадацитиниб,
- Олокизумаб,
- Левилимаб,
- Тоцилизумаб,
- Сарилумаб,
- Канакинумаб,
- Анакинра,
- Метилпреднизолон,
- Дексаметазон,
- Гидрокортизон,
- Будесонид.
В список возможных к назначению антикоагулянтов для лечения COVID-19 у взрослых включены:
1) антикоагулянты для парентерального введения:
- нефракционированный гепарин: нефракционированный гепарин.
- низкомолекулярные гепарины: далтепарин натрия, надропарин кальция, эноксапарин натрия, парнапарин натрия, бемипарин натрия.
- синтетические антикоагулянты: фондапаринукс натрия.
- ривароксабан, апиксабан, дабигатрана этексилат.
2) пероральные антикоагулянты:
По процедуре регистрации препаратов, предназначенных для применения в условиях угрозы возникновения, возникновения и ликвидации чрезвычайных ситуаций зарегистрирован ряд препаратов и вакцин, рекомендованных к применению для лечения и профилактики новой коронавирусной инфекции (COVID-19).
* См. Версия 18 (26.10.2023) — Временные методические рекомендации профилактики, диагностики и лечения новой коронавирусной инфекции (COVID-19) — Минздрав России.
Классификация
-
Код МКБ 10
Информация о действующем веществе Бамланивимаб + Этесевимаб предназначена для медицинских и фармацевтических специалистов, исключительно в справочных целях. Инструкция не предназначена для замены профессиональной медицинской консультации, диагностики или лечения. Содержащаяся здесь информация может меняться с течением времени. Наиболее точные сведения о применении препаратов, содержащих активное вещество Бамланивимаб + Этесевимаб, содержатся в инструкции производителя, прилагаемой к упаковке.
В журнале NEJM опубликованы результаты рандомизированного плацебо- контролируемого исследования III фазы BLAZE-1, в котором «коктейль» из антител к SARS-CoV2 (бамланивимаб и этесевимаб, производство Eli Lilly) сравнивался с плацебо. В исследование было включено 1035 пациентов, получающих лечение в амбулаторных условиях, у которых тяжесть COVID-19 была оценена как легкая или умеренная, но присутствовали дополнительные факторы риска прогрессирования заболевания (такие, например, как диабет, ожирение 2 степени и более, возраст 65+, ХБП и проч.). Критериям включения соответствовали пациенты, у которых с момента положительного ПЦР теста прошло не более 3 суток. Таким пациентам однократно проводилась инфузия бамланивимаба и этесевимаба (по 2800 мг каждого) или плацебо. Медиана времени от появления симптомов до введения исследуемого препарата или плацебо составила 4 дня. Длительность наблюдения составила 29 дней.
В группе введения антител смертность и потребность в госпитализации составила 2,1%, в группе плацебо – 7% (снижение частоты первичной конечной точки составило 70%, р<0.001). В группе терапии антителами не умерло ни одного человека, в группе плацебо умерло 10 человек (по мнению исследователей, в 9 случаев причиной смерти стал непосредственно COVID-19).
Бамланивимаб и этесивимаб характеризовались приемлемым спектром безопасности, частота побочных эффектов не различалась между группой антител и группой плацебо. Ни у одного пациента, включенного в исследование, лечение не было прекращено в связи с развитием неблагоприятных событий.
Кроме того, в группе бамланивимаба и этесивимаба на 7-е сутки степень вирусовыделения была достоверно ниже, положительный ПЦР регистрировался у 9,8% пациентов группы антител, 29,5% группы плацебо. Т.о., помимо улучшения прогноза у заболевших пациентов, данный вид лечения снижает риск дальнейшего распространения SARS-CoV2.
По материалам:
Dougan M, Nirula A, Azizad M, et al. Bamlanivimab plus Etesevimab in Mild or Moderate Covid-19. N Engl J Med. 2021 Jul 14 doi: 10.1056/NEJMoa2102685. Epub ahead of print. PMID: 34260849
https://pubmed.ncbi.nlm.nih.gov/
Текст: Шахматова О.О.
RECENT MAJOR CHANGES
|
Revised 01/2022 |
|
Revised 12/2021 |
|
Revised 12/2021, 08/2021, 05/2021, and 03/2021 |
|
Revised 12/2021 |
|
Revised 12/2021 |
|
Revised 09/2021 |
|
Revised 09/2021 |
|
Revised 08/2021 |
|
Revised 08/2021 |
|
Revised 08/2021 |
|
Revised 08/2021 |
|
Revised 05/2021 |
|
Revised 05/2021 |
|
Revised 05/2021 |
|
Revised 05/2021 |
Bamlanivimab and etesevimab have been authorized by FDA for the emergency uses described above.
Bamlanivimab and etesevimab are not FDA-approved for these uses.
Bamlanivimab and etesevimab are authorized only for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of bamlanivimab and etesevimab under section 564(b)(1) of the Act, 21 U.S.C. § 360bbb-3(b)(1), unless the authorization is terminated or revoked sooner.
Dosing
See Full Fact Sheet for Healthcare Providers for information on dosing [see Dosage and Administration (2)].
Preparation and Administration
See Full Fact Sheet for Healthcare Providers for information on preparation and administration [see Dose Preparation and Administration (2.4)].
Under this EUA, single-dose vials may be used to prepare more than one pediatric dose; in addition, pediatric doses do not need to be diluted for patients <18 years and weighing <40kg.
Storage and Handling
Refrigerate unopened vials at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light. Do not freeze, shake, or expose to direct light.
FDA has authorized an extension to the shelf-life (i.e., expiration date) of both bamlanivimab and etesevimab following a thorough review of data submitted by Eli Lilly and Company. The extension applies to all unopened vials of bamlanivimab and etesevimab that have been held in accordance with storage conditions. Confirm the shelf-life of unopened vials of bamlanivimab and etesevimab by batch number at the FDA EUA website under the Drug and Biological Therapeutic Products bamlanivimab and etesevimab. This site includes a complete listing of extended expiration dates by batch number. If the batch number on the vial/carton is not included in this listing, the product is labeled with the correct expiration date.
Warnings
There are limited clinical data available for bamlanivimab and etesevimab. Serious and unexpected adverse events may occur that have not been previously reported with use of bamlanivimab and etesevimab together.
Hypersensitivity Including Anaphylaxis and Infusion-Related Reactions
Serious hypersensitivity reactions, including anaphylaxis, have been observed with administration of bamlanivimab and etesevimab. If signs and symptoms of a clinically significant hypersensitivity reaction or anaphylaxis occur, immediately discontinue administration and initiate appropriate medications and/or supportive therapy.
Infusion-related reactions, occurring during the infusion and up to 24 hours after the infusion, have been observed with administration of bamlanivimab and etesevimab together. These reactions may be severe or life threatening.
Signs and symptoms of infusion related reactions may include:
- fever, difficulty breathing, reduced oxygen saturation, chills, fatigue, arrhythmia (e.g., atrial fibrillation, sinus tachycardia, bradycardia), chest pain or discomfort, weakness, altered mental status, nausea, headache, bronchospasm, hypotension, hypertension, angioedema, throat irritation, rash including urticaria, pruritus, myalgia, vasovagal reactions (e.g., pre-syncope, syncope), dizziness and diaphoresis.
Consider slowing or stopping the infusion and administer appropriate medications and/or supportive care if an infusion-related reaction occurs.
Hypersensitivity reactions occurring more than 24 hours after the infusion have also been reported with the use of bamlanivimab and etesevimab under Emergency Use Authorization.
Clinical Worsening After Bamlanivimab and Etesevimab Administration
Clinical worsening of COVID-19 after administration of bamlanivimab and etesevimab together has been reported and may include signs or symptoms of fever, hypoxia or increased respiratory difficulty, arrhythmia (e.g., atrial fibrillation, sinus tachycardia, bradycardia), fatigue, and altered mental status. Some of these events required hospitalization. It is not known if these events were related to bamlanivimab and etesevimab use or were due to progression of COVID-19.
Limitations of Benefit and Potential for Risk in Patients with Severe COVID-19
Treatment with bamlanivimab and etesevimab has not been studied in patients hospitalized due to COVID-19. Monoclonal antibodies, such as bamlanivimab and etesevimab, may be associated with worse clinical outcomes when administered to hospitalized patients with COVID-19 requiring high flow oxygen or mechanical ventilation. Therefore, [see Limitations of Authorized Use (1.1)]:
- Bamlanivimab and etesevimab are not authorized for use in patients 2 years and older who are hospitalized due to COVID-192,
- Bamlanivimab and etesevimab are not authorized for use in patients, regardless of age, who:
- require oxygen therapy and/or respiratory support due to COVID-19, OR
- require an increase in baseline oxygen flow rate and/or respiratory support due to COVID-19 and are on chronic oxygen therapy and/or respiratory support due to underlying non-COVID-19 related comorbidity.
Side Effects
Adverse events have been reported with bamlanivimab and etesevimab [see Full EUA Prescribing Information, Overall Safety Summary (6.1)].
Additional adverse events associated with bamlanivimab and etesevimab, some of which may be serious, may become apparent with more widespread use.
INSTRUCTIONS FOR HEALTHCARE PROVIDERS
As the healthcare provider, you must communicate to your patient or parent/caregiver, as age appropriate, information consistent with the “Fact Sheet for Patients, Parents and Caregivers” (and provide a copy of the Fact Sheet) prior to the patient receiving bamlanivimab and etesevimab, including:
- FDA has authorized the emergency use of bamlanivimab and etesevimab administered together for the treatment of mild to moderate COVID-19 in adults and pediatric patients, including neonates, with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death [see Limitations of Authorized Use (1.1)].
- FDA has authorized the emergency use of bamlanivimab and etesevimab administered together in adults and pediatric individuals, including neonates, for post-exposure prophylaxis of COVID-19 in individuals who are at high risk for progression to severe COVID-19, including hospitalization or death, and are:
- not fully vaccinated3 or who are not expected to mount an adequate immune response to complete SARS-CoV-2 vaccination (for example, individuals with immunocompromising conditions including those taking immunosuppressive medications4) and
- have been exposed to an individual infected with SARS-CoV-2 consistent with close contact criteria per Centers for Disease Control and Prevention (CDC)5 or
- who are at high risk of exposure to an individual infected with SARS-CoV-2 because of occurrence of SARS-CoV-2 infection in other individuals in the same institutional setting (for example, nursing homes, prisons) [see Limitations of Authorized Use (1.2)].
- not fully vaccinated3 or who are not expected to mount an adequate immune response to complete SARS-CoV-2 vaccination (for example, individuals with immunocompromising conditions including those taking immunosuppressive medications4) and
- The patient or parent/caregiver has the option to accept or refuse bamlanivimab and etesevimab.
- The significant known and potential risks and benefits of bamlanivimab and etesevimab, and the extent to which such potential risks and benefits are unknown.
- Information on available alternative treatments and the risks and benefits of those alternatives, including clinical trials.
- Patients treated with bamlanivimab and etesevimab together should continue to self-isolate and use infection control measures (e.g., wear mask, isolate, social distance, avoid sharing personal items, clean and disinfect “high touch” surfaces, and frequent handwashing) according to CDC guidelines.
For information on clinical trials that are testing the use of bamlanivimab and etesevimab together for COVID-19, please see www.clinicaltrials.gov.
MANDATORY REQUIREMENTS FOR BAMLANIVIMAB AND ETESEVIMAB ADMINISTRATION UNDER EMERGENCY USE AUTHORIZATION:
In order to mitigate the risks of using these unapproved products and to optimize the potential benefit of bamlanivimab and etesevimab under this EUA, the following items are required. Use of bamlanivimab and etesevimab under this EUA is limited to the following (all requirements must be met):
- Treatment of mild to moderate COVID-19 in adults and pediatric patients, including neonates, with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death [see Limitations of Authorized Use (1.1)].
- Post-exposure prophylaxis of COVID-19 in adults and pediatric individuals, including neonates, who are at high risk for progression to severe COVID-19, including hospitalization or death, and are:
- not fully vaccinated3 or who are not expected to mount an adequate immune response to complete SARS-CoV-2 vaccination (for example, individuals with immunocompromising conditions including those taking immunosuppressive medications4) and
- have been exposed to an individual infected with SARS-CoV-2 consistent with close contact criteria per Centers for Disease Control and Prevention (CDC)5 or
- who are at high risk of exposure to an individual infected with SARS-CoV-2 because of occurrence of SARS-CoV-2 infection in other individuals in the same institutional setting (for example, nursing homes, prisons) [see Limitations of Authorized Use (1.2)].
- not fully vaccinated3 or who are not expected to mount an adequate immune response to complete SARS-CoV-2 vaccination (for example, individuals with immunocompromising conditions including those taking immunosuppressive medications4) and
- As the healthcare provider, communicate to your patient or parent/caregiver, as age appropriate, information consistent with the “Fact Sheet for Patients, Parents and Caregivers” prior to the patient receiving bamlanivimab and etesevimab. Healthcare providers (to the extent practicable given the circumstances of the emergency) must document in the patient’s medical record that the patient/caregiver has been:
- Given the “Fact Sheet for Patients, Parents and Caregivers”,
- Informed of alternatives to receiving authorized bamlanivimab and etesevimab, and
- Informed that bamlanivimab and etesevimab are unapproved drugs that are authorized for use under this Emergency Use Authorization.
- Patients with known hypersensitivity to any ingredient of bamlanivimab or etesevimab must not receive bamlanivimab and etesevimab.
- The prescribing health care provider and/or the provider’s designee is/are responsible for mandatory reporting of all medication errors and serious adverse events* potentially related to bamlanivimab and etesevimab treatment within 7 calendar days from the onset of the event. The reports must include unique identifiers and the words “bamlanivimab and etesevimab use for COVID-19 under Emergency Use Authorization (EUA)” in the description section of the report.
- Submit adverse event reports to FDA MedWatch using one of the following methods:
- Complete and submit the report online: www.fda.gov/medwatch/report.htm, or
- Complete and submit a postage-paid FDA Form 3500 (https://www.fda.gov/media/76299/download) and return by:
- Mail to MedWatch, 5600 Fishers Lane, Rockville, MD 20852-9787, or
- Fax (1-800-FDA-0178), or
- Call 1-800-FDA-1088 to request a reporting form.
- Submitted reports must include in the field name, “Describe Event, Problem, or Product Use/Medication Error” the statement “bamlanivimab and etesevimab use for COVID-19 under Emergency Use Authorization (EUA).”
*Serious Adverse Events are defined as:
- death;
- a life-threatening adverse event;
- inpatient hospitalization or prolongation of existing hospitalization;
- a persistent or significant incapacity or substantial disruption of the ability to conduct normal life functions;
- a congenital anomaly/birth defect;
- a medical or surgical intervention to prevent death, a life-threatening event, hospitalization, disability, or congenital anomaly.
- Submit adverse event reports to FDA MedWatch using one of the following methods:
- The prescribing health care provider and/or the provider’s designee is/are to provide mandatory responses to requests from FDA for information about adverse events and medication errors following receipt of bamlanivimab and etesevimab.
- OTHER REPORTING REQUIREMENTS
- Healthcare facilities and providers must report therapeutics information and utilization data through HHS Protect, Teletracking or National Healthcare Safety Network (NHSN) as directed by the U.S. Department of Health and Human Services.
- In addition, please provide a copy of all FDA MedWatch forms to:
Eli Lilly and Company, Global Patient Safety
Fax: 1-317-277-0853
E-mail: mailindata_gsmtindy@lilly.com
Or call Eli Lilly and Company at 1-855-LillyC19 (1-855-545-5921) to report adverse events.
APPROVED AVAILABLE ALTERNATIVES
Veklury (remdesivir) is FDA-approved for the treatment of COVID-19 in adults and pediatric patients (12 years of age and older weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing, who are not hospitalized and have mild-to-moderate COVID-19, and who are at high risk for progression to severe COVID-19, including hospitalization or death. Veklury is administered via intravenous infusion for a total treatment duration of 3 days.
Although Veklury is an approved alternative treatment of mild-to-moderate COVID-19 in adults and pediatric patients (12 years of age and older weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death, FDA does not consider Veklury to be an adequate alternative to bamlanivimab and etesevimab for this authorized use because it may not be feasible or practical for certain patients (e.g., it requires a 3-day treatment duration).6
There is no adequate, approved and available alternative to bamlanivimab and etesevimab administered together for post-exposure prophylaxis of COVID-19 in adult and pediatric individuals, including neonates, who are at high risk for progression to severe COVID-19, including hospitalization or death, and are:
- not fully vaccinated3 or who are not expected to mount an adequate immune response to complete SARS-CoV-2 vaccination (for example, individuals with immunocompromising conditions including those taking immunosuppressive medications4) and
- have been exposed to an individual infected with SARS-CoV-2 consistent with close contact criteria per Centers for Disease Control and Prevention (CDC)5 or
- who are at high risk of exposure to an individual infected with SARS-CoV-2 because of occurrence of SARS-CoV-2 infection in other individuals in the same institutional setting (for example, nursing homes, prisons) [see Limitations of Authorized Use (1.2)].
Additional information on COVID-19 therapies can be found at https://www.cdc.gov/coronavirus/2019-ncov/index.html. The health care provider should visit https://clinicaltrials.gov/ to determine whether the patient may be eligible for enrollment in a clinical trial.
- 6
- Additionally, the approval for Veklury does not cover certain pediatric patients for whom bamlanivimab and etesevimab administered together is authorized (e.g., patients less than 12 years of age).
AUTHORITY FOR ISSUANCE OF THE EUA
The Secretary of the Department of Health and Human Services (HHS) has declared a public health emergency that justifies the emergency use of drugs and biological products during the COVID-19 pandemic. FDA has issued this EUA, requested by Eli Lilly and Company for the unapproved products bamlanivimab and etesevimab administered together for the treatment of mild to moderate COVID-19 in adults and pediatric patients, including neonates, with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death.5
FDA has also issued this EUA, requested by Eli Lilly and Company for the unapproved products bamlanivimab and etesevimab administered together in adults and pediatric individuals, including neonates, for post-exposure prophylaxis of COVID-19 in individuals who are at high risk of progression to severe COVID-19, including hospitalization or death, and are:
- not fully vaccinated3 or who are not expected to mount an adequate immune response to complete SARS-CoV-2 vaccination (for example, individuals with immunocompromising conditions including those taking immunosuppressive medications4) and
- have been exposed to an individual infected with SARS-CoV-2 consistent with close contact criteria per Centers for Disease Control and Prevention (CDC)5 or
- who are at high risk of exposure to an individual infected with SARS-CoV-2 because of occurrence of SARS-CoV-2 infection in other individuals in the same institutional setting (for example, nursing homes, prisons) [see Limitations of Authorized Use (1.2)].
Although limited scientific information is available, based on the totality of the scientific evidence available to date, it is reasonable to believe that bamlanivimab and etesevimab administered together may be effective for the treatment of mild to moderate COVID-19 or for post-exposure prophylaxis of COVID-19 in individuals as specified in this Fact Sheet. You may be contacted and asked to provide information to help with the assessment of the use of the product during this emergency.
This EUA for bamlanivimab and etesevimab will end when the Secretary determines that the circumstances justifying the EUA no longer exist or when there is a change in the approval status of the product such that an EUA is no longer needed.
As a health care provider, you must comply with the mandatory requirements of the EUA (see above).
CONTACT INFORMATION
For additional information visit
www.LillyAntibody.com
If you have questions, please contact
1-855-LillyC19 (1-855-545-5921)
| END SHORT VERSION FACT SHEET Long Version Begins on Next Page |
FULL EUA PRESCRIBING INFORMATION
| FULL EUA PRESCRIBING INFORMATION: CONTENTS* |
11.2 Lactation | ||
| 1 AUTHORIZED USE |
11.3 Pediatric Use | ||
| 1.1 Treatment | 11.4 Geriatric Use | ||
| 1.2 Post-Exposure Prophylaxis | 11.5 Renal Impairment | ||
| 2 DOSAGE AND ADMINISTRATION |
11.6 Hepatic Impairment | ||
| 2.1 Patient Selection | 11.7 Other Specific Populations | ||
| 2.2 Dosage | 12 OVERDOSAGE |
||
| 2.3 Dosage Adjustment in Specific Populations | 13 DESCRIPTION |
||
| 2.4 Dose Preparation and Administration | 14 CLINICAL PHARMACOLOGY |
||
| 3 DOSAGE FORMS AND STRENGTHS |
14.1 Mechanism of Action | ||
| 4 CONTRAINDICATIONS |
14.2 Pharmacodynamics | ||
| 5 WARNINGS AND PRECAUTIONS |
14.3 Pharmacokinetics | ||
| 5.1 Hypersensitivity Including Anaphylaxis and Infusion-Related Reactions | 15 MICROBIOLOGY/RESISTANCE INFORMATION |
||
| 5.2 Clinical Worsening After Bamlanivimab and Etesevimab Administration | 16 NONCLINICAL TOXICOLOGY |
||
| 5.3 Limitations of Benefit and Potential for Risk in Patients with Severe COVID-19 | 17 ANIMAL PHARMACOLOGIC AND EFFICACY DATA |
||
| 6 OVERALL SAFETY SUMMARY |
18 CLINICAL TRIAL RESULTS AND SUPPORTING DATA FOR EUA | ||
| 6.1 Clinical Trials Experience | 18.1 Treatment of Mild to Moderate COVID-19 (BLAZE-1) | ||
| 7 PATIENT MONITORING RECOMMENDATIONS |
18.2 Post-Exposure Prophylaxis of COVID-19 (BLAZE-2) |
||
| 8 ADVERSE REACTIONS AND MEDICATION ERRORS REPORTING REQUIREMENTS AND INSTRUCTIONS |
19 HOW SUPPLIED/STORAGE AND HANDLING |
||
| 9 OTHER REPORTING REQUIREMENTS |
20 PATIENT COUNSELING INFORMATION |
||
| 10 DRUG INTERACTIONS |
21 CONTACT INFORMATION |
||
| 11 USE IN SPECIFIC POPULATIONS |
* Sections or subsections omitted from the full prescribing information are not listed. | ||
| 11.1 Pregnancy |
1 AUTHORIZED USE
1.1 TREATMENT
Bamlanivimab and etesevimab administered together are authorized for use under an EUA for the treatment of mild to moderate coronavirus disease 2019 (COVID-19) in adults and pediatric patients, including neonates, with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death.
Limitations of Authorized Use
- Bamlanivimab and etesevimab are not authorized for treatment of mild to moderate COVID-19 in geographic regions where infection is likely to have been caused by a non-susceptible SARS-CoV-2 variant based on available information including variant susceptibility to these drugs and regional variant frequency.
- FDA’s determination and any updates will be available at: https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization#coviddrugs.1
- Bamlanivimab and etesevimab are not authorized for use in patients 2 years and older who are hospitalized due to COVID-19.2
- Bamlanivimab and etesevimab are not authorized for use in patients, regardless of age, who:
- require oxygen therapy and/or respiratory support due to COVID-19, OR
- require an increase in baseline oxygen flow rate and/or respiratory support due to COVID-19 and are on chronic oxygen therapy and/or respiratory support due to underlying non-COVID-19 related comorbidity.
- Treatment with bamlanivimab and etesevimab has not been studied in patients hospitalized due to COVID-19. Monoclonal antibodies, such as bamlanivimab and etesevimab, may be associated with worse clinical outcomes when administered to hospitalized patients with COVID-19 requiring high flow oxygen or mechanical ventilation [see Warnings and Precautions (5.3)].
1.2 POST-EXPOSURE PROPHYLAXIS
Bamlanivimab and etesevimab administered together are authorized for use under an EUA for post-exposure prophylaxis of COVID-19 in adults and pediatric individuals, including neonates, who are at high risk for progression to severe COVID-19, including hospitalization or death, and are:
- not fully vaccinated3 or who are not expected to mount an adequate immune response to complete SARS-CoV-2 vaccination (for example, individuals with immunocompromising conditions including those taking immunosuppressive medications4) and
- have been exposed to an individual infected with SARS-CoV-2 consistent with close contact criteria per Centers for Disease Control and Prevention (CDC)5 or
- who are at high risk of exposure to an individual infected with SARS-CoV-2 because of occurrence of SARS-CoV-2 infection in other individuals in the same institutional setting (for example, nursing homes, prisons).
Limitations of Authorized Use
- Bamlanivimab and etesevimab are not authorized for post-exposure prophylaxis of COVID-19 in geographic regions where exposure is likely to have been to a non-susceptible SARS-CoV-2 variant based on available information including variant susceptibility to these drugs and regional variant frequency.
- FDA’s determination and any updates will be available at: https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization#coviddrugs.1
- Post-exposure prophylaxis with bamlanivimab and etesevimab is not a substitute for vaccination against COVID-19.
- Bamlanivimab and etesevimab are not authorized for pre-exposure prophylaxis for prevention of COVID-19.
2. Bamlanivimab and Etesevimab Injection Dosage and Administration
2.1 Patient Selection
The following medical conditions or other factors may place adults and pediatric patients, including neonates, at higher risk for progression to severe COVID-19:
- Older age (for example age ≥65 years of age)
- <1 year old
- Obesity or being overweight
- Pregnancy
- Chronic kidney disease
- Diabetes
- Immunosuppressive disease or immunosuppressive treatment
- Cardiovascular disease (including congenital heart disease) or hypertension
- Chronic lung diseases (for example, chronic obstructive pulmonary disease, asthma [moderate-to-severe], interstitial lung disease, cystic fibrosis and pulmonary hypertension)
- Sickle cell disease
- Neurodevelopmental disorders (for example, cerebral palsy) or other conditions that confer medical complexity (for example, genetic or metabolic syndromes and severe congenital anomalies)
- Having a medical-related technological dependence (for example, tracheostomy, gastrostomy, or positive pressure ventilation (not related to COVID-19))
Other medical conditions or factors (for example, race or ethnicity) may also place individual patients at high risk for progression to severe COVID-19 and authorization of bamlanivimab and etesevimab under the EUA is not limited to the medical conditions or factors listed above. For additional information on medical conditions and factors associated with increased risk for progression to severe COVID-19, see the CDC website: https://www.cdc.gov/coronavirus/2019-ncov/need-extra-precautions/people-with-medical-conditions.html. Healthcare providers should consider the benefit-risk for an individual patient.
2.2 Dosage
Treatment:
The dosage in adults (18 years and older) and pediatric patients (<18 years and weighing at least 40 kg) is bamlanivimab 700 mg and etesevimab 1,400 mg. The dosage for pediatric patients weighing less than 40 kg will vary depending on body weight:
- >20 kg to <40 kg: 350 mg bamlanivimab and 700 mg etesevimab
- >12 kg to 20 kg: 175 mg bamlanivimab and 350 mg etesevimab
- 1 kg to 12 kg: 12 mg/kg bamlanivimab and 24 mg/kg etesevimab
The recommended dosing regimen for pediatric patients ≤12 kg is predicted based on pharmacokinetic modeling and simulation [see Clinical Pharmacology (14.3)]. The youngest participant in the pediatric clinical trial for treatment was 10 months of age and weighed 8.6 kg [see Use in Specific Populations (11.3) and Clinical Trials and Supporting Data for EUA (18.1)].
For treatment of COVID-19, bamlanivimab and etesevimab should be administered together as soon as possible after positive results of direct SARS-CoV-2 viral testing and within 10 days of symptom onset.
Post-Exposure Prophylaxis:
The dosage in adults (18 years and older) and pediatric individuals (<18 years and weighing at least 40 kg) is 700 mg bamlanivimab and 1,400 mg etesevimab administered together as a single intravenous infusion. The dosage for pediatric individuals weighing less than 40 kg will vary depending on body weight:
- >20 kg to <40 kg: 350 mg bamlanivimab and 700 mg etesevimab
- >12 kg to 20 kg: 175 mg bamlanivimab and 350 mg etesevimab
- 1 kg to 12 kg: 12 mg/kg bamlanivimab and 24 mg/kg etesevimab
The recommended dosing regimen for pediatric patients ≤12 kg is predicted based on pharmacokinetic modeling and simulation [see Clinical Pharmacology (14.3)]. The youngest participant in the pediatric clinical trial for treatment was 10 months of age and weighed 8.6 kg [see Use in Specific Populations (11.3) and Clinical Trials and Supporting Data for EUA (18.1)]. Children were not enrolled in the post-exposure prophylaxis trial, BLAZE-2 [see Clinical Trials and Supporting Data for EUA (18.2)].
For post-exposure prophylaxis, bamlanivimab and etesevimab should be given together as soon as possible following exposure to SARS-CoV-2.
Under this EUA, bamlanivimab and etesevimab must be administered together as a single intravenous infusion.
2.3 Dosage Adjustment in Specific Populations
Pregnancy or Lactation
No dosage adjustment is recommended in pregnant or lactating women [see Use in Specific Populations (11.1, 11.2)].
Pediatric Use
No dosage adjustment is recommended in pediatric patients <18 years who weigh at least 40 kg. For pediatric patients weighing less than 40 kg, dosage adjustment on the basis of body weight is required [see Dosage and Administration (2.4)]. The recommended dosing regimen for pediatric patients ≤12 kg is predicted based on pharmacokinetic modeling and simulation [see Clinical Pharmacology (14.3)]. The youngest participant in the pediatric clinical trial for treatment was 10 months of age and weighed 8.6 kg [see Use in Specific Populations (11.3) and Clinical Trials and Supporting Data for EUA (18.1)]. Children were not enrolled in the post-exposure prophylaxis trial, BLAZE-2 [see Clinical Trials and Supporting Data for EUA (18.2)].
Geriatric Use
No dosage adjustment is recommended in geriatric patients [see Use in Specific Populations (11.4)].
Renal Impairment
No dosage adjustment is recommended in patients with renal impairment [see Use in Specific Populations (11.5)].
Hepatic Impairment
No dosage adjustment is recommended in patients with mild hepatic impairment. Bamlanivimab and etesevimab has not been studied in patients with moderate or severe hepatic impairment [see Use in Specific Populations (11.6)].
2.4 Dose Preparation and Administration
General Information
- Bamlanivimab and etesevimab solution for infusion should be prepared by a qualified healthcare professional using aseptic technique.
- Bamlanivimab and etesevimab are supplied in individual vials but are administered together.
- Inspect bamlanivimab and etesevimab vials visually for particulate matter and discoloration. Bamlanivimab and etesevimab are clear to opalescent and colorless to slightly yellow to slightly brown solutions.
- The prepared infusion solution should not be administered simultaneously with any other medication. The compatibility of bamlanivimab and etesevimab injection with IV solutions and medications other than 0.9% Sodium Chloride Injection is not known.
- If the infusion must be discontinued due to an infusion reaction, discard any unused product.
- The use of closed system transfer devices (CSTDs), elastomeric pumps, and pneumatic transport with bamlanivimab and etesevimab has not been studied.
- Clinically monitor patients during administration and observe patients for at least 1 hour after infusion is complete.
IV Infusion in
Adults (≥18 years regardless of weight)
and
Pediatric Patients (<18 years and weighing at least 40 kg)
Materials Needed
- 1 bamlanivimab vial (700 mg/20 mL)
- 2 etesevimab vials (700 mg/20 mL)
- 1 polyvinyl chloride (PVC) or polyethylene (PE)-line PVC, sterile prefilled infusion bag containing 0.9% Sodium Chloride Injection (sizes 50 mL to 250 mL)
- 1 PVC or PE-lined PVC infusion set
- 1 in-line or add-on 0.2/0.22 micron polyethersulfone (PES) filter
- 0.9% Sodium Chloride for flushing tubing
Preparation
- Remove 1 bamlanivimab vial and 2 etesevimab vials from refrigerated storage and allow to equilibrate to room temperature for approximately 20 minutes before preparation. Do not expose to direct heat. Do not shake the vials. Inspect vials.
- Withdraw 20 mL from one bamlanivimab vial and 40 mL from two etesevimab vials and inject all 60 mL into a prefilled infusion bag containing 0.9% Sodium Chloride (see Table 1).
- Discard any product remaining in the vials.
- Gently invert the bag by hand approximately 10 times to mix. Do not shake.
|
a 700 mg of bamlanivimab and 1,400 mg of etesevimab are added to the same infusion bag and administered together as a single intravenous infusion. |
||
|
b The minimum infusion time for patients weighing at least 40 kg and less than 50 kg who are administered bamlanivimab and etesevimab diluted in a 250-mL prefilled 0.9% Sodium Chloride infusion bag must be extended to at least 70 minutes to reduce endotoxin load. |
||
| Druga: Add 20 mL of bamlanivimab (1 vial) and 40 mL of etesevimab (2 vials) for a total of 60 mL to a prefilled infusion bag and administer as instructed below | ||
| Size of Prefilled 0.9% Sodium Chloride Infusion Bag | Maximum Infusion Rate | Minimum Infusion Time |
| 50 mL | 310 mL/hr | 21 minutes |
| 100 mL | 310 mL/hr | 31 minutes |
| 150 mL | 310 mL/hr | 41 minutes |
| 250 mL For patients weighing at least 50 kg |
310 mL/hr | 60 minutes |
| 250 mLb For patients weighing ≥40 kg and <50 kg |
266 mL/hr | 70 minutes |
Administration
- These products are preservative-free and therefore, the diluted infusion solution should be administered immediately.
- If immediate administration is not possible, store the diluted infusion solution for up to 24 hours at refrigerated temperature (2°C to 8°C [36°F to 46°F]) and up to 7 hours at room temperature (20°C to 25°C [68°F to 77°F]) including infusion time. If refrigerated, allow the infusion solution to equilibrate to room temperature for approximately 20 minutes prior to administration.
- Attach the infusion set to the IV bag. Use of in-line or add-on 0.2/0.22 micron polyethersulfone (PES) filter is strongly recommended.
- Prime the infusion set.
- Administer the entire infusion solution in the bag via pump or gravity according to the size of infusion bag used (see Table 1). Due to potential overfill of prefilled saline bags, the entire infusion solution in the bag should be administered to avoid underdosage.
- Once infusion is complete, flush the tubing with 0.9% Sodium Chloride to ensure delivery of the required dose.
IV Infusion in
Pediatric Patients (<18 years and weighing <40 kg)
Materials Needed
| IV bag | Syringe Pump |
| 1 bamlanivimab vial (700 mg/20 mL) | 1 bamlanivimab vial (700 mg/20 mL) |
| 1 etesevimab vial (700 mg/20 mL) | 1 etesevimab vial (700 mg/20 mL) |
| 1 sterile, empty 50-mL PVC or PE-lined PVC infusion bag | 1 disposable syringe |
| 1 PVC or PE-lined PVC Infusion set | 1 syringe extension set |
| 1 in-line or add-on 0.2/0.22 micron PES filter | 1 syringe pump |
| 0.9% Sodium Chloride for flushing | 0.9% Sodium Chloride for flushing |
Under this EUA, single-dose vials may be used to prepare more than one pediatric dose; in addition, pediatric doses do not need to be diluted for patients <18 years and weighing <40 kg.
Preparation
- Remove bamlanivimab and etesevimab vials from refrigerated storage and allow to equilibrate to room temperature for approximately 20 minutes before preparation. Do not expose to direct heat. Do not shake vials. Inspect vials.
- Withdraw appropriate amounts of bamlanivimab and etesevimab from vials based on body weight and inject into the empty infusion bag or draw into a disposable syringe (see Table 2).
- Multiple doses of bamlanivimab and etesevimab may be prepared from each product vial (see the storage conditions specified below). Prepare all infusion bags or syringes at the same time. Appropriately label any prepared doses including the patient weight and dose, and time of preparation to minimize risk of medication errors, particularly in cases where multiple doses are prepared simultaneously.
- Discard any product remaining in the vials after all doses have been prepared.
- Gently invert the infusion bag or syringe to mix the contents. Do not shake or vigorously agitate.
|
a Amount of BAM (as mL) and amount of ETE (as mL) for patients weighing up to 12 kg are calculated and rounded to one decimal place. |
||||
| Body Weight | BAM/ETE dose (mg) |
Amount of BAM (as mL)a |
Amount of ETE (as mL)a |
Maximum Infusion Rate |
| >20 kg to <40 kg | 350 mg / 700 mg | 10 mL | 20 mL | 1.88 mL/min |
| >12 kg to 20 kg | 175 mg / 350 mg | 5 mL | 10 mL | 0.94 mL/min |
| >11 kg to 12 kg | 138 mg / 276 mg | 3.9 mL | 7.9 mL | 0.74 mL/min |
| >10 kg to 11 kg | 126 mg / 252 mg | 3.6 mL | 7.2 mL | 0.68 mL/min |
| >9 kg to 10 kg | 114 mg / 228 mg | 3.3 mL | 6.5 mL | 0.61 mL/min |
| >8 kg to 9 kg | 102 mg / 204 mg | 2.9 mL | 5.8 mL | 0.54 mL/min |
| >7 kg to 8 kg | 90 mg / 180 mg | 2.6 mL | 5.1 mL | 0.48 mL/min |
| >6 kg to 7 kg | 78 mg / 156 mg | 2.2 mL | 4.5 mL | 0.42 mL/min |
| >5 kg to 6 kg | 66 mg / 132 mg | 1.9 mL | 3.8 mL | 0.36 mL/min |
| >4 kg to 5 kg | 54 mg / 108 mg | 1.5 mL | 3.1 mL | 0.29 mL/min |
| >3 kg to 4 kg | 42 mg / 84 mg | 1.2 mL | 2.4 mL | 0.23 mL/min |
| >2 kg to 3 kg | 30 mg / 60 mg | 0.9 mL | 1.7 mL | 0.16 mL/min |
| >1.5 kg to 2 kg | 21 mg / 42 mg | 0.6 mL | 1.2 mL | 0.11 mL/min |
| 1 kg to 1.5 kg | 15 mg / 30 mg | 0.4 mL | 0.9 mL | 0.08 mL/min |
Administration
- These products are preservative-free and therefore, the infusion solution should be administered immediately.
- If immediate administration is not possible, store the infusion solution for up to 24 hours at refrigerated temperature (2°C to 8°C [36°F to 46°F]) and up to 7 hours at room temperature (20°C to 25°C [68°F to 77°F]) including infusion time. If refrigerated, allow the infusion solution to equilibrate to room temperature for approximately 20 minutes prior to administration.
- IV bag:
- Attach the infusion set to the IV bag. Use of in-line or add-on 0.2/0.22 micron polyethersulfone (PES) filter is strongly recommended.
- Prime the infusion set.
- Administer the entire infusion solution in the bag via pump or gravity over at least 16 minutes (see Table 2).
- Once infusion is complete, flush the tubing with 0.9% Sodium Chloride to ensure delivery of the required dose.
- Syringe Pump:
- Administer the entire contents of the syringe via syringe pump over at least 16 minutes (see Table 2).
- After the entire contents of the syringe have been administered, flush the extension set with 0.9% Sodium Chloride to ensure delivery of the required dose.
3. Dosage Forms and Strengths
Bamlanivimab is a sterile, preservative-free, clear to opalescent and colorless to slightly yellow to slightly brown solution available as:
- Injection: 700 mg/20 mL (35 mg/mL) in a single-dose* vial.
Etesevimab is a sterile, preservative-free, clear to opalescent and colorless to slightly yellow to slightly brown solution available as:
- Injection: 700 mg/20 mL (35 mg/mL) in a single-dose* vial.
* Under this EUA, single-dose vials may be used to prepare more than one pediatric dose.
4. Contraindications
None.
5. Warnings and Precautions
There are limited clinical data available for bamlanivimab and etesevimab. Serious and unexpected adverse events may occur that have not been previously reported with use of bamlanivimab and etesevimab together.
5.1 Hypersensitivity Including Anaphylaxis and Infusion-Related Reactions
Serious hypersensitivity reactions, including anaphylaxis, have been observed with administration of bamlanivimab and etesevimab. If signs and symptoms of a clinically significant hypersensitivity reaction or anaphylaxis occur, immediately discontinue administration and initiate appropriate medications and/or supportive care.
Infusion-related reactions, occurring during the infusion and up to 24 hours after the infusion, have been observed with administration of bamlanivimab and etesevimab together. These reactions may be severe or life threatening.
Signs and symptoms of infusion related reactions may include [see Overall Safety Summary (6.1)]:
- fever, difficulty breathing, reduced oxygen saturation, chills, fatigue, arrhythmia (e.g., atrial fibrillation, sinus tachycardia, bradycardia), chest pain or discomfort, weakness, altered mental status, nausea, headache, bronchospasm, hypotension, hypertension, angioedema, throat irritation, rash including urticaria, pruritus, myalgia, vasovagal reactions (e.g., pre-syncope, syncope), dizziness and diaphoresis.
Consider slowing or stopping the infusion and administer appropriate medications and/or supportive care if an infusion-related reaction occurs.
Hypersensitivity reactions occurring more than 24 hours after the infusion have also been reported with the use of bamlanivimab and etesevimab under Emergency Use Authorization.
5.2 Clinical Worsening After Bamlanivimab and Etesevimab Administration
Clinical worsening of COVID-19 after administration of bamlanivimab and etesevimab together has been reported and may include signs or symptoms of fever, hypoxia or increased respiratory difficulty, arrhythmia (e.g., atrial fibrillation, sinus tachycardia, bradycardia), fatigue, and altered mental status. Some of these events required hospitalization. It is not known if these events were related to bamlanivimab and etesevimab use or were due to progression of COVID-19.
5.3 Limitations of Benefit and Potential for Risk in Patients with Severe COVID-19
Treatment with bamlanivimab and etesevimab has not been studied in patients hospitalized due to COVID-19. Monoclonal antibodies, such bamlanivimab and etesevimab, may be associated with worse clinical outcomes when administered to hospitalized patients with COVID-19 requiring high flow oxygen or mechanical ventilation. Therefore,
- Bamlanivimab and etesevimab are not authorized for use in patients 2 years and older who are hospitalized due to COVID-192,
- Bamlanivimab and etesevimab are not authorized for use in patients, regardless of age, who:
- require oxygen therapy and/or respiratory support due to COVID-19, OR
- require an increase in baseline oxygen flow rate and/or respiratory support due to COVID-19 and are on chronic oxygen therapy and/or respiratory support due to underlying non-COVID-19 related comorbidity [see Limitations of Authorized Use (1.1)].
6 OVERALL SAFETY SUMMARY
6.1 Clinical Trials Experience
Adults (≥18 Years) and Pediatric Patients (<18 Years and weighing at least 40 kg)
The safety of bamlanivimab administered with etesevimab is primarily based on exposure of approximately 1,400 ambulatory (non-hospitalized) subjects who received doses of bamlanivimab and etesevimab together, at the recommended dose or higher, in BLAZE-1 and BLAZE-4. BLAZE-1 is a Phase 2/3, randomized, double-blind, placebo-controlled clinical trial studying bamlanivimab and etesevimab administered together for the treatment of subjects with mild to moderate COVID-19. Thirty-four pediatric patients (ages 12 to <18 years and weighing at least 40 kg) were included in the Phase 3 portion of BLAZE-1 (14 received placebo, 14 received the authorized dose or a higher dose for their age, and 6 received a lower dose than authorized for their age). In the Phase 3 portion of the trial, enrolled participants had at least one risk factor for the development of severe COVID-19 illness. BLAZE-4 is a Phase 2, randomized, double-blind, placebo-controlled clinical trial studying bamlanivimab and etesevimab for the treatment of subjects with mild to moderate COVID-19. Subjects ≥65 years old or with BMI ≥35 were excluded from enrollment. In clinical trials, approximately 4,000 subjects have received bamlanivimab (either alone or with etesevimab) at doses ranging from 700 to 7,000 mg. Bamlanivimab and etesevimab at the authorized doses of 700 mg and 1,400 mg have been administered together to approximately 800 subjects in clinical trials [see Clinical Pharmacology (14.2)].
The following adverse reactions (i.e., adverse events assessed as causally related) have been observed in those who have received bamlanivimab and etesevimab together at the authorized dose or higher [see Warnings and Precautions (5.1)]:
- anaphylaxis (n=1, 0.07%)
- infusion-related reactions (n=16, 1.1%)
In the case of anaphylaxis and serious infusion-related reactions, all infusions were stopped, and treatment was administered. One case required epinephrine. All events resolved.
The most common treatment-emergent adverse events in the bamlanivimab and etesevimab treatment group in BLAZE-1 and BLAZE-4 included nausea, dizziness, and pruritus. No treatment-emergent adverse events occurred in more than 1% of participants and the rates were comparable in the treatment and placebo groups.
Pediatric Patients (Birth to <18 Years)
In addition to the 34 pediatric patients (ages 12 to <18 and weighing at least 40 kg) enrolled in the Phase 3 portion of BLAZE-1, an open-label pediatric addendum to BLAZE-1 enrolled 40 patients aged 12 to <18, 36 aged 6 to <12, 10 aged 2 to <6, and 5 birth to <2 for a total of 125 pediatric patients. All pediatric patients had at least one risk factor for the development of severe COVID-19 illness. Pediatric patients weighing 8.6 kg to <40 kg received doses of bamlanivimab and etesevimab that were adjusted for their body weight, to achieve comparable exposures as adults and adolescents receiving the authorized dosage of bamlanivimab 700 mg and etesevimab 1,400 mg, respectively. The adverse drug reaction profile in pediatric patients is consistent with the established profile.
7 PATIENT MONITORING RECOMMENDATIONS
Clinically monitor patients during administration and observe patients for at least 1 hour after infusion is complete [see Warnings and Precautions (5.1) and Overall Safety Summary (6.1)].
8. Adverse Reactions/Side Effects
Clinical trials evaluating the safety of bamlanivimab and etesevimab are ongoing [see Overall Safety Summary (6)].
Completion of FDA MedWatch Form to report all medication errors and serious adverse events* occurring during bamlanivimab and etesevimab use and considered to be potentially related to bamlanivimab and etesevimab is mandatory and must be done by the prescribing healthcare provider and/or the provider’s designee. These adverse events must be reported within 7 calendar days from the onset of the event:
*Serious adverse events are defined as:
- death;
- a life-threatening adverse event;
- inpatient hospitalization or prolongation of existing hospitalization;
- a persistent or significant incapacity or substantial disruption of the ability to conduct normal life functions;
- a congenital anomaly/birth defect;
- a medical or surgical intervention to prevent death, a life-threatening event, hospitalization, disability, or congenital anomaly.
If a serious and unexpected adverse event occurs and appears to be associated with the use of bamlanivimab and etesevimab under this EUA, the prescribing healthcare provider and/or the provider’s designee must complete and submit a MedWatch form to FDA using one of the following methods:
- Complete and submit the report online: www.fda.gov/medwatch/report.htm, or
- Complete and submit a postage-paid FDA Form 3500 (https://www.fda.gov/media/76299/download) and return by:
- Mail to MedWatch, 5600 Fishers Lane, Rockville, MD 20852-9787, or
- Fax (1-800-FDA- 0178), or
- Call 1-800-FDA-1088 to request a reporting form
IMPORTANT: When reporting adverse events or medication errors to MedWatch, please complete the entire form with detailed information. It is important that the information reported to FDA be as detailed and complete as possible. Information that must be included:
- Patient demographics (e.g., patient initials, date of birth)
- Pertinent medical history
- Pertinent details regarding adverse events and course of illness
- Concomitant medications
- Timing of adverse event(s) in relationship to administration of bamlanivimab and etesevimab
- Pertinent laboratory and virology information
- Outcome of the event and any additional follow-up information if it is available at the time of the MedWatch report. Subsequent reporting of follow-up information should be completed if additional details become available.
The following steps are highlighted to provide the necessary information for safety tracking:
- In section A, box 1, provide the patient’s initials in the Patient Identifier
- In section A, box 2, provide the patient’s date of birth
- In section B, box 5, description of the event:
- Write “bamlanivimab and etesevimab use for COVID-19 under Emergency Use Authorization (EUA)” as the first line
- Provide a detailed report of medication error and/or adverse event. It is important to provide detailed information regarding the patient and adverse event/medication error for ongoing safety evaluation of this unapproved drug. Please see information to include listed above.
- In section G, box 1, name and address:
- Provide the name and contact information of the prescribing healthcare provider or institutional designee who is responsible for the report.
- Provide the address of the treating institution (NOT the healthcare provider’s office address).
9 OTHER REPORTING REQUIREMENTS
- Healthcare facilities and providers must report therapeutics information and utilization data through HHS Protect, Teletracking or National Healthcare Safety Network (NHSN) as directed by the U.S. Department of Health and Human Services.
- In addition, please provide a copy of all FDA MedWatch forms to:
Eli Lilly and Company, Global Patient Safety
Fax: 1-317-277-0853
E-mail: mailindata_gsmtindy@lilly.com
Or call Eli Lilly and Company at 1-855-LillyC19 (1-855-545-5921) to report adverse events.
10. Drug Interactions
Bamlanivimab and etesevimab are not renally excreted or metabolized by cytochrome P450 enzymes; therefore, interactions with concomitant medications that are renally excreted or that are substrates, inducers, or inhibitors of cytochrome P450 enzymes are unlikely.
11. Use In Specific Populations
11.1 Pregnancy
Risk Summary
There are insufficient data to evaluate a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. Bamlanivimab and etesevimab should only be used during pregnancy if the potential benefit outweighs the potential risk for the mother and the fetus. There are maternal and fetal risks associated with untreated COVID-19 in pregnancy (see Clinical Considerations).
Nonclinical reproductive toxicity studies have not been performed with bamlanivimab or etesevimab. In tissue cross reactivity studies using human fetal tissues, no binding of clinical concern was detected for etesevimab or bamlanivimab. Human immunoglobulin G1 (IgG1) antibodies are known to cross the placental barrier; therefore, bamlanivimab and etesevimab have the potential to be transferred from the mother to the developing fetus. It is unknown whether the potential transfer of bamlanivimab or etesevimab provides any treatment benefit or risk to the developing fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo-Fetal Risk
COVID-19 in pregnancy is associated with adverse maternal and fetal outcomes, including preeclampsia, eclampsia, preterm birth, premature rupture of membranes, venous thromboembolic disease, and fetal death.
11.2 Lactation
Risk Summary
There are no available data on the presence of bamlanivimab or etesevimab in human or animal milk, the effects on the breastfed infant, or the effects on milk production. Maternal IgG is known to be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for bamlanivimab and etesevimab and any potential adverse effects on the breastfed child from bamlanivimab and etesevimab or from the underlying maternal condition. Breastfeeding individuals with COVID-19 should follow practices according to clinical guidelines to avoid exposing the infant to COVID-19.
11.3 Pediatric Use
Bamlanivimab and etesevimab administered together are authorized for the treatment of mild to moderate COVID-19 and post-exposure prophylaxis for prevention of COVID-19 in pediatric patients, including neonates [see Authorized Use (1)]. Given the similar course of COVID-19, the authorization of bamlanivimab and etesevimab for treatment and post-exposure prophylaxis in younger pediatric patients, including neonates, is supported by safety and efficacy data in adolescents and adults, together with additional pharmacokinetic and safety data from the clinical trial in pediatric patients studying bamlanivimab and etesevimab for the treatment of mild to moderate COVID-19.
Use of bamlanivimab and etesevimab in pediatric patients is based on analyses of data from BLAZE-1 in subjects aged 10 months to 18 years of age [see Clinical Pharmacology (14.3) and Clinical Trials and Supporting Data for EUA (18.1)]. No dosage adjustment is recommended in pediatric patients 12-18 years of age who weigh at least 40 kg. Pediatric patients weighing less than 40 kg should be dosed on the basis of body weight [see Dosage and Administration (2.2, 2.4)]. The recommended dosing regimen for pediatric patients ≤12 kg is predicted based on pharmacokinetic modeling and simulation [see Clinical Pharmacology (14.3)]. The youngest participant in the pediatric clinical trial for treatment was 10 months of age and weighed 8.6 kg [see Clinical Trials and Supporting Data for EUA (18.1)]. Safety in pediatric patients was similar to what was observed in adults [see Clinical Trial Experience (6.1)]. Children were not enrolled in the post-exposure prophylaxis trial, BLAZE-2 [see Clinical Trials and Supporting Data for EUA (18.2)].
11.4 Geriatric Use
Of the 1141 patients receiving bamlanivimab and etesevimab in BLAZE-1, 30% were 65 years of age and older and 10% were 75 years of age and older. Based on population PK analyses, there is no difference in PK of bamlanivimab or etesevimab in geriatric patients compared to younger patients [see Clinical Trial Results and Supporting Data for EUA (18.1)].
11.5 Renal Impairment
Bamlanivimab and etesevimab are not eliminated intact in the urine, thus renal impairment is not expected to affect the exposure of bamlanivimab or etesevimab.
11.6 Hepatic Impairment
Based on population PK analysis, there is no difference in PK of bamlanivimab or etesevimab in patients with mild hepatic impairment compared to patients with normal hepatic function. Bamlanivimab and etesevimab have not been studied in patients with moderate or severe hepatic impairment.
11.7 Other Specific Populations
Based on population PK analysis, the PK of bamlanivimab and etesevimab was not affected by sex, race, or disease severity. Body weight had no clinically relevant effect on the PK of bamlanivimab and etesevimab in adults with COVID-19 over the body weight range of 41 kg to 173 kg.
12. Overdosage
Doses up to 7,000 mg of bamlanivimab (10 times the authorized dose of bamlanivimab for adults [≥18 years] and pediatric patients [<18 years weighing at least 40 kg]) or 7,000 mg of etesevimab (5 times the authorized dose of etesevimab for adults [≥18 years] and pediatric patients [<18 years weighing at least 40 kg]) have been administered in clinical trials without dose-limiting toxicity. Treatment of overdose with bamlanivimab and etesevimab should consist of general supportive measures including monitoring of vital signs and observation of the clinical status of the patient. There is no specific antidote for overdose with either bamlanivimab or etesevimab.
13. Bamlanivimab and Etesevimab Injection Description
Bamlanivimab
Bamlanivimab is a human immunoglobulin G-1 (IgG1 variant) monoclonal antibody consisting of 2 identical light chain polypeptides composed of 214 amino acids each and 2 identical heavy chain polypeptides composed of 455 amino acids produced by a Chinese Hamster Ovary (CHO) cell line and molecular weight of 146 kDa.
Bamlanivimab injection is a sterile, preservative-free, clear to opalescent and colorless to slightly yellow to slightly brown solution in a vial for intravenous infusion.
Each mL contains 35 mg of bamlanivimab, and L-histidine (0.4 mg), L-histidine hydrochloride monohydrate (0.6 mg), sodium chloride (2.9 mg), sucrose (60 mg), polysorbate 80 (0.5 mg), and Water for Injection. The bamlanivimab solution has a pH range of 5.5-6.5.
Etesevimab
Etesevimab is a human IgG1 variant monoclonal antibody (mAb) consisting of 2 identical light chain polypeptides composed of 216 amino acids each and 2 identical heavy chain polypeptides composed of 449 amino acids produced by a Chinese Hamster Ovary (CHO) cell line and molecular weight of 145 kDa.
Etesevimab injection is a sterile, preservative-free, clear to opalescent and colorless to slightly yellow to slightly brown solution in a vial for intravenous infusion.
Each mL contains 35 mg of etesevimab, L-histidine (1.55 mg), L-histidine hydrochloride monohydrate (2.10 mg), sucrose (80.4 mg), polysorbate 80 (0.5 mg), and Water for injection. The etesevimab solution has a pH range of 5.5.-6.5.
14. Bamlanivimab and Etesevimab Injection — Clinical Pharmacology
14.1 Mechanism of Action
Bamlanivimab is a recombinant neutralizing human IgG1ϰ monoclonal antibody (mAb) to the spike protein of SARS-CoV-2 and is unmodified in the Fc region. Bamlanivimab binds the spike protein with a dissociation constant KD = 0.071 nM and blocks spike protein attachment to the human ACE2 receptor with an IC50 value of 0.17 nM (0.025 μg/mL).
Etesevimab is a recombinant neutralizing human IgG1ϰ mAb to the spike protein of SARS-CoV-2, with amino acid substitutions in the Fc region (L234A, L235A) to reduce effector function. Etesevimab binds the spike protein with a dissociation constant KD = 6.45 nM and blocks spike protein attachment to the human ACE2 receptor with an IC50 value of 0.32 nM (0.046 μg/mL).
Bamlanivimab and etesevimab bind to different but overlapping epitopes in the receptor binding domain (RBD) of the S-protein. Using both antibodies together is expected to reduce the risk of viral resistance.
14.2 Pharmacodynamics
A flat exposure-response relationship for efficacy was identified for bamlanivimab and etesevimab administered together within the dose range of 700 mg bamlanivimab and 1,400 mg etesevimab to 2,800 mg bamlanivimab and 2,800 mg etesevimab (4 and 2 times the authorized dose, respectively), based on clinical data and pharmacokinetic/pharmacodynamic modeling.
For post-exposure prophylaxis of COVID-19, a dose of 700 mg bamlanivimab and 1,400 mg etesevimab was supported based on clinical data and pharmacokinetic/pharmacodynamic modeling.
14.3 Pharmacokinetics
A summary of PK parameters of bamlanivimab and etesevimab following administration of a single dose of 700 mg bamlanivimab and 1,400 mg etesevimab is provided in Table 3. There is no change in PK of bamlanivimab or etesevimab administered alone or together suggesting there is no interaction between the two antibodies. There were no differences in PK of etesevimab between mild/moderate ambulatory participants and healthy participants.
|
Abbreviations: CV = coefficient of variation; Cmax = maximum concentration; AUCinf = area under the concentration versus time curve from zero to infinity; Vss = steady-state volume of distribution. |
|||
|
a N = number of subjects simulated using the PK model. |
|||
|
b The number of subjects for Vss, half-life, and clearance are based on a population PK model that included bamlanivimab doses up to 7,000 mg and etesevimab doses up to 2,800 mg. |
|||
| N | BAM (700 mg) |
ETE (1400 mg) |
|
| Systemic Exposure | |||
| Geometric Mean (%CV) Cmax, mcg/mL | 270 | 187 (41.7) | 422 (41.2) |
| Geometric Mean (%CV) Cday 29, mcg/mL | 311 BAM; 320 ETE | 25.7 (42.9) | 116 (38.1) |
| Median (5th,95th percentile) Cweek 8, mcg/mL | 1000a | 10.1 (3.59, 22.9) | 58.3 (26.8, 117) |
| Geometric Mean (%CV) AUCinf, mcg day/mL | 499 | 2500 (28.0) | 10600 (29.9) |
| Distribution | |||
| Geometric Mean (%CV) Vss (L) | 1899 BAM; 1498 ETEb | 6.59 (24.9) | 5.78 (24.7) |
| Elimination | |||
| Geometric Mean (%CV) Elimination Half-Life (day) | 1899 BAM; 1498 ETEb | 20.9 (17.3) | 32.6 (21.7) |
| Geometric Mean (%CV) Clearance (L/day) | 1899 BAM; 1498 ETEb | 0.274 (31.5) | 0.134 (32.5) |
Bamlanivimab and etesevimab are expected to be degraded into small peptides and component amino acids via catabolic pathways in the same manner as endogenous IgG antibodies.
Special Populations:
The PK profiles of bamlanivimab and etesevimab were not affected by age, sex, race, or disease severity based on a population PK analysis. Body weight had no clinically relevant effect on the PK of bamlanivimab or etesevimab in adults with COVID-19 over the body weight range of 41 kg to 173 kg [see Use in Specific Populations (11.4, 11.7)].
Pediatric population
The PK of bamlanivimab and etesevimab has been evaluated in 88 pediatric patients <18 years who received weight-based dosing [see Dosage and Administration (2.2)]. The data show that weight-based dosing in pediatric patients provides comparable plasma exposures to those observed in adults who received bamlanivimab 700 mg and etesevimab 1,400 mg. No dosage adjustment is recommended in pediatric patients <18 years who weigh at least 40 kg. Pediatric patients weighing less than 40 kg should be dosed on the basis of body weight [see Dosage and Administration (2.2, 2.4)]. The recommended dosing regimen for pediatric patients ≤12 kg is predicted to result in similar exposures when compared to exposures achieved in adults receiving bamlanivimab 700 mg and etesevimab 1,400 mg based on pharmacokinetic modeling and simulation. The youngest participant in the pediatric treatment trial was 10 months of age and weighed 8.6 kg [see Clinical Trials and Supporting Data for EUA (18.1)].
|
Abbreviations: CV = coefficient of variation; Cmax = maximum concentration; AUCinf = area under the concentration versus time curve from zero to infinity. |
||||
| Body Weight | ≥40 kg | >20 to <40 kg | >12 to ≤20 kg | ≤12 kg |
| BAM / ETE Dose | 700 mg / 1400 mg | 350 mg / 700 mg | 175 mg / 350 mg | 15 mg/kg / 30 mg/kg |
| BAM: Geometric Mean (%CV) [n] | ||||
| Cmax, mcg/mL | 235 (51.0) [52] | 239 (39.1) [16] | 243 (66.0) [7] | 371 (9.8) [2] |
| Cday 29, mcg/mL | 26.8 (50.2) [34] | 26.1 (32.5) [8] | 23.0 (53.0) [3] | [0] |
| AUCinf, mcg day/mL | 2760 (30.7) [66] | 2780 (25.7) [20] | 2430 (28.4) [9] | 3000 (19.1) [3] |
| ETE: Geometric Mean (%CV) [n] | ||||
| Cmax, mcg/mL | 508 (50.6) [50] | 444 (26.6) [14] | 444 (64.9) [7] | 831 (16.8) [2] |
| Cday 29, mcg/mL | 133 (46.8) [34] | 138 (29.5) [8] | 125 (51.5) [3] | [0] |
| AUCinf, mcg day/mL | 12900 (32.4) [66] | 12400 (23.2) [20] | 11300 (29.6) [9] | 13500 (13.0) [3] |
Patients with renal impairment
Bamlanivimab and etesevimab are not eliminated intact in the urine. Renal impairment is not expected to impact the PK of bamlanivimab and etesevimab, since mAbs with molecular weight >69 kDa are known not to undergo renal elimination. Similarly, dialysis is not expected to impact the PK of bamlanivimab and etesevimab [see Use in Specific Populations (11.5)].
Patients with hepatic impairment
Based on population PK analysis, there is no significant difference in PK of bamlanivimab or etesevimab in patients with mild hepatic impairment compared to patients with normal hepatic function. Bamlanivimab and etesevimab have not been studied in patients with moderate or severe hepatic impairment [see Use in Specific Populations (11.6)].
Drug interactions:
Bamlanivimab and etesevimab are not renally excreted or metabolized by cytochrome P450 enzymes; therefore, interactions with concomitant medications that are renally excreted or that are substrates, inducers, or inhibitors of cytochrome P450 enzymes are unlikely.
15 MICROBIOLOGY/RESISTANCE INFORMATION
Antiviral Activity
The cell culture neutralization activity of bamlanivimab and of etesevimab against SARS-CoV-2 was measured in a dose-response model quantifying plaque reduction using cultured Vero E6 cells. Bamlanivimab, etesevimab and a 1:1 (weight/weight) ratio of bamlanivimab and etesevimab together neutralized the USA/WA/1/2020 isolate of SARS-CoV-2 with estimated EC50 values = 0.14 nM (0.02 μg/mL), 0.97 nM (0.14 μg/mL) and 0.14 nM (0.02 μg/mL), respectively.
Bamlanivimab demonstrated antibody-dependent cell-mediated cytotoxicity on reporter Jurkat cells expressing FcγRIIIa following engagement with target cells expressing spike protein. Bamlanivimab did not elicit complement-dependent cytotoxicity activity in cell-based assays.
Etesevimab did not demonstrate detectable antibody-dependent cell-mediated cytotoxicity on Jurkat reporter cells expressing FcγRIIIa. Etesevimab did not elicit complement-dependent cytotoxicity activity in cell-based assays.
Antibody Dependent Enhancement (ADE) of Infection
The risk that bamlanivimab and etesevimab could mediate viral uptake and replication by immune cells was studied in THP-1 and Raji cell lines and primary human macrophages. In general, experiments with bamlanivimab, with etesevimab, and with bamlanivimab and etesevimab together did not demonstrate productive viral infection in immune cells exposed to SARS-CoV-2 at concentrations of mAb(s) down to at least 100-fold below the respective EC50 value(s).
Antiviral Resistance
There is a potential risk of treatment failure due to the development of viral variants that are resistant to bamlanivimab and/or etesevimab (Table 5). There are other authorized treatments available and healthcare providers should choose an authorized therapeutic option with activity against circulating variants in their state, territory, or US jurisdiction. Variant frequency data for states, territories, and US jurisdictions can be accessed on the following CDC website: https://www.cdc.gov/coronavirus/2019-ncov/cases-updates/variant-proportions.html.
Resistant variants were identified using directed evolution of the spike protein and serial passage in cell culture of SARS-CoV-2 in the presence of bamlanivimab or etesevimab individually. Resistant variants were not identified when bamlanivimab and etesevimab were tested together using the same methodology. Viral variants identified in these studies that had reduced susceptibility to bamlanivimab included spike protein amino acid substitutions E484D/K/Q, F490S, Q493R, and S494P, and variants that had reduced susceptibility to etesevimab included substitutions K417N, D420N, and N460K/S/T/Y. Neutralization assays using SARS-CoV-2 and vesicular stomatitis virus (VSV) virus-like particles (VLP) pseudotyped with variant SARS-CoV-2 spike protein confirmed reductions in susceptibility to the selecting antibody. Retention of susceptibility to the other antibody alone was observed, with the exception of the E484D and Q493R substitution. All variants maintained susceptibility to bamlanivimab and etesevimab together, with the exception of those with E484D, E484K, E484Q, and Q493R substitutions, which had reduced susceptibility of 145-fold, 24-fold, 17-fold, and 1,054-fold, respectively in a pseudotyped VLP assay.
Evaluation of susceptibility of variants identified through global surveillance in subjects treated with bamlanivimab and etesevimab is ongoing. Pseudotyped VLP evaluation of amino acid substitutions identified in global surveillance showed that the V483A substitution reduced susceptibility to bamlanivimab 48-fold, but activity was maintained with etesevimab, and with bamlanivimab and etesevimab together. N501Y and N501T substitutions reduced susceptibility to etesevimab approximately 5-fold and 20-fold, respectively. Activity against variants with N501Y or N501T substitutions was maintained with bamlanivimab alone, and with bamlanivimab and etesevimab together.
Bamlanivimab and etesevimab together retained activity against a SARS-CoV-2 B.1.1.7 lineage (Alpha; UK origin) virus and related pseudotyped VLPs expressing the spike protein found in the B.1.1.7 variant (Tables 5 and 6). SARS-CoV-2 B.1.351 lineage (Beta; South Africa origin) virus and related pseudotyped VLPs expressing spike proteins from B.1.351 lineage or substitutions K417N + E484K + N501Y found in this lineage had reduced susceptibility to bamlanivimab and etesevimab together of >324, 431-fold or >45-fold, respectively. Pseudotyped VLPs expressing spike protein from the P.1 lineage (Gamma; Brazil origin) or K417T + E484K + N501Y found in the P.1 lineage had reduced susceptibility to bamlanivimab and etesevimab together of 252-fold or >3,351-fold, respectively.
Bamlanivimab and etesevimab together and etesevimab alone retained activity against SARS-CoV-2 B.1.617.2 lineage (Delta; India origin) virus and related pseudotyped VLPs, but bamlanivimab alone had reduced activity (>1,136 and >1,868-fold, respectively). Bamlanivimab and etesevimab are expected to retain activity against B.1.617.2 sublineage AY.3 (India origin). B.1.617.2 sublineages AY.1/AY.2 (India origin) have an additional K417N substitution; pseudotyped VLPs expressing AY.1/AY.2 related spike sequence had a reduced susceptibility to bamlanivimab and etesevimab together of 1,235-fold. SARS-CoV-2 recombinant virus containing the L452R substitution present in B.1.427/B.1.429 lineages (Epsilon; USA [California] origin) and pseudotyped VLPs expressing the full-length spike protein or the L452R substitution found in this lineage showed reduced susceptibility to bamlanivimab and etesevimab together of 11-fold, 9-fold or 5-fold, respectively. Pseudotyped VLPs expressing spike protein from the B.1.617.1 lineage (Kappa; India origin) showed reduced susceptibility to bamlanivimab and etesevimab together of 6-fold; for this variant, susceptibility to etesevimab alone was maintained, but not to bamlanivimab alone (>1,030-fold reduction). Bamlanivimab and etesevimab together and etesevimab alone retained activity against pseudotyped VLPs expressing the full-length spike protein from the C.37 lineage (Lambda; Peru origin), but bamlanivimab alone had reduced activity (>2,112-fold reduction). Pseudotyped VLPs expressing spike protein from the B.1.621 lineage (Mu; Colombia origin) show reduced susceptibility to bamlanivimab and etesevimab together of 116-fold, due to susceptibility reductions to bamlanivimab (>1,863-fold) and etesevimab (17-fold) alone. Pseudotyped VLPs expressing the spike protein from the B.1.1.529/BA.1 lineage (Omicron; South Africa origin) show reduced susceptibility to bamlanivimab alone (>1,465-fold), etesevimab alone (>616-fold), and bamlanivimab and etesevimab together (>2,938-fold).
|
a Key substitutions occurring in the receptor binding domain of spike protein are listed. Pseudoviruses containing the full-length spike protein reflective of the consensus sequence for each of the variant lineages were tested. |
||||
|
b No change: <5-fold reduction in susceptibility. |
||||
|
c Bamlanivimab and etesevimab together are unlikely to be active against variants from this lineage. |
||||
|
d Etesevimab retains activity against this variant. |
||||
|
e Isolates of the B.1.526 lineage harbor several spike protein amino acid substitutions, and not all isolates contain the E484K substitution (as of February 2021). |
||||
| Lineage with Spike Protein Substitution | Country First Identified | WHO Nomenclature | Key Substitutions Testeda | Fold Reduction in Susceptibility |
| B.1.1.7 | UK | Alpha | N501Y | no changeb |
| B.1.351 | South Africa | Beta | K417N + E484K + N501Y | 431c |
| P.1 | Brazil | Gamma | K417T + E484K + N501Y | 252c |
| B.1.617.2/AY.3 | India | Delta | L452R + T478K | no changeb |
| AY.1/AY.2 (B.1.617.2 sublineages) |
India | Delta [+K417N] | L452R + T478K + K417N | 1,235c |
| B.1.427/B.1.429 | USA (California) | Epsilon | L452R | 9d |
| B.1.526e | USA (New York) | Iota | E484K | 30 |
| B.1.617.1 | India | Kappa | L452R + E484Q | 6d |
| C.37 | Peru | Lambda | L452Q + F490S | no changeb |
| B.1.621 | Colombia | Mu | R346K + E484K + N501Y | 116c |
| B.1.1.529/BA.1 | South Africa | Omicron | G339D + S371L + S373P + S375F + K417N + N440K + G446S + S477N + T478K + E484A + Q493R + G493S + Q498R + N501Y + Y505H | >2,938c |
|
a The B.1.1.7 variant was assessed using cell culture-expanded virus isolates and tested using an immunofluorescence based microneutralization assay and by plaque reduction assay; B.1.351 and B.1.617.2 variants were assessed using cell culture-expanded virus isolates and tested using a plaque reduction assay; the B.1.526/E484K and B.1.427/B.1.429/L452R substitutions were assessed using recombinant SARS-CoV-2 (USA/WA/1/2020 isolate with E484K or L452R) and tested using a plaque reduction assay. |
||||
|
b Key substitutions occurring in receptor binding domain of spike protein which are associated with each lineage. |
||||
|
c No change: <5-fold reduction in susceptibility. |
||||
|
d Isolates of the B.1.526 lineage harbor several spike protein amino acid substitutions, and not all isolates contain the E484K substitution (as of February 2021). This assay was conducted using recombinant SARS-CoV-2 with the E484K substitution only. |
||||
| Lineage with Spike Protein Substitution | Country First Identified | WHO Nomenclature | Key Substitutions Testedb | Fold Reduction in Susceptibility |
| B.1.1.7 | UK | Alpha | N501Y | no changec |
| B.1.351 | South Africa | Beta | K417N, E484K, N501Y | >324 |
| B.1.617.2/AY.3 | India | Delta | L452R, T478K | no changec |
| B.1.427/B.1.429 | USA (California) | Epsilon | L452R | 11 |
| B.1.526d | USA (New York) | Iota | E484K | 11 |
Due to the large reduction of pseudotyped VLP neutralization activity of both bamlanivimab and etesevimab against the substitutions in B.1.351 (Beta; South Africa origin), P.1 (Gamma; Brazil origin), AY.1/AY.2 (Delta [+K417N]; India origin), B.1.621 (Mu; Colombia origin), and B.1.1.529/BA.1 (Omicron; South Africa origin), it is unlikely that bamlanivimab and etesevimab together will be active against these variants.
It is unclear how small reductions in susceptibility to bamlanivimab and etesevimab seen in authentic or recombinant SARS-CoV-2 or pseudotyped VLP assays correlate with clinical outcomes.
In authentic SARS-CoV-2 assays, bamlanivimab and etesevimab together retained activity against variants of B.1.1.7 (Alpha) and B.1.617.2/AY.3 (Delta) lineages (Table 6), although bamlanivimab alone had reduced activity to B.1.617.2/AY.3 (Delta) in this assay (>1,136-fold). SARS-CoV-2 (USA/WA/1/2020 isolate) engineered to express the E484K substitution present in the B.1.526 lineage (Iota; USA [New York] origin) or the L452R substitution present in the B.1.427/B.1.429 lineage (Epsilon; USA [California] origin) showed reduced susceptibility to bamlanivimab and etesevimab together of 11-fold. Susceptibility to etesevimab alone was maintained for both isolates, but not to bamlanivimab alone (>833-fold and >1,460-fold reduction for E484K and L452R viruses, respectively). Available nonclinical and clinical PK data indicate that etesevimab at the authorized dose may retain activity against the B.1.526 variant clinically, although only very limited data are currently available from patients infected with this variant in clinical trials. Preliminary clinical evidence indicates that the administration of bamlanivimab and etesevimab together result in similar viral load reductions in participants infected with the L452R variant (Epsilon; USA [California] origin) as observed in those who were infected with bamlanivimab-sensitive strains. Of the 134 participants infected with the L452R variant at baseline in the Phase 3 portion of BLAZE-1, 3 of the 50 individuals treated with placebo (6%) and 1 of the 84 participants treated with bamlanivimab 700 mg and etesevimab 1,400 mg (1%) were hospitalized (p=0.15).
Genotypic and phenotypic testing are ongoing to monitor for potential bamlanivimab- and etesevimab-resistance associated spike variations in clinical trials. Analysis of baseline samples show that 8.4% (188/2246) of clinical trial patients were infected with viral variants containing single amino acid substitutions at positions associated with reduced susceptibility to either bamlanivimab or etesevimab as predicted by pseudotyped VLP or authentic SARS-CoV-2 neutralization assays. No patients were infected with a variant that was predicted to have reduced susceptibility to both bamlanivimab and etesevimab by these assessments.
Patient samples were also analyzed for treatment-emergent viral variants, defined as variants with single amino acid substitutions at positions that had reduced susceptibility to either bamlanivimab or etesevimab present at an allele fraction of ≥15%.
- In the Phase 3 portion of BLAZE-1, treatment-emergent variants were observed in 9.0% (42/467) of patients treated with bamlanivimab 2,800 mg and etesevimab 2,800 mg together, in 5.3% (21/394) of patients treated with bamlanivimab 700 mg and etesevimab 1,400 mg together, and in 4.0% (27/674) of patients treated with placebo. The majority of these were only detected at one time point in the sequential series with 0.9% (4/467), 1.0% (4/394), and 0.3% (2/674) of patients having multiple instances of detection in the bamlanivimab 2,800 mg and etesevimab 2,800 mg together, bamlanivimab 700 mg and etesevimab 1,400 mg together, and placebo groups, respectively.
- In patients treated with bamlanivimab and etesevimab together, substitutions detected in one or more patients included ones with reduced susceptibility (≥5-fold) to bamlanivimab only: L452R/W, E484K, G485V, F490L, and S494P; and ones with reduced susceptibility to etesevimab only: D405G/Y, K417N, D420N/Y, N460H/I/T, A475S/V, Y489H, and N501I/Y. While these variants had reduced susceptibility to either bamlanivimab OR etesevimab compared to wild-type in a pseudotyped VSV VLP or authentic virus assay they still retained susceptibility to the other antibody in the combination.
- There were also observations of variants with reduced susceptibility (≥5-fold) to both bamlanivimab and etesevimab and to bamlanivimab + etesevimab tested together: E484D (n=1; 145-fold reduction to bamlanivimab + etesevimab tested together at a molar ratio of 1:2), Q493K/R (n=9; 584-fold and 1,054-fold reduction to bamlanivimab + etesevimab tested together at a molar ratio of 1:2 for Q493K and Q493R, respectively) out of a total of 861 patients treated with bamlanivimab and etesevimab together.
- In a subgroup of participants infected with virus harboring L452R substitution found in the B.1.427/B.1.429 (Epsilon) lineage, a S459P treatment-emergent substitution was identified in one subject. Concurrent L452R+S459P substitutions conferred a 1,656-fold reduction in susceptibility to bamlanivimab + etesevimab together (1:2 molar ratio).
- Additional treatment-emergent substitutions in patients treated with bamlanivimab and etesevimab together, with no phenotypic data, include D405del, D420G, C480R, G485D, S494L, and P499L. The impact of these substitutions on susceptibility is not currently known.
- In a subgroup of 53 pediatric subjects who were infected with a B.1.617.2 (Delta)-related variant, which has reduced susceptibility to bamlanivimab (>1,136-fold), the following treatment-emergent substitutions with reduced susceptibility to etesevimab were detected: D420A (n=2), N460T (n=1), N460Y (n=1). Three of these four subjects had high viral load (>5.27 log10) on Day 7.
- Additional treatment-emergent substitutions with no phenotypic data detected in other pediatric subjects who were infected with a B.1.617.2 (Delta)-related variant at an allele fraction of ≥50% included: F347C, V401L, G431S and I434V.
It is possible that bamlanivimab and etesevimab resistance-associated variants could have cross-resistance to other mAbs targeting the receptor binding domain of SARS-CoV-2. The clinical impact is not known.
Immune Response Attenuation
There is a theoretical risk that antibody administration may attenuate the endogenous immune response to SARS-CoV-2 and make patients more susceptible to re-infection.
16. Nonclinical Toxicology
Carcinogenesis, mutagenesis, and reproductive toxicology studies with bamlanivimab or etesevimab have not been conducted.
In toxicology studies, bamlanivimab and etesevimab had no adverse effects when administered intravenously to rats and monkeys, respectively. Non-adverse increases in neutrophils were observed in rats dosed with bamlanivimab.
In tissue cross reactivity studies using human adult and fetal tissues, no binding of clinical concern was detected for bamlanivimab or etesevimab.
17 ANIMAL PHARMACOLOGIC AND EFFICACY DATA
Antiviral Activity In Vivo
Prophylactic administration of bamlanivimab to female Rhesus macaques (n=3 or 4 per group) resulted in 1 to 4 log10 decreases in viral genomic RNA and viral replication (sub-genomic RNA) in bronchoalveolar lavage samples relative to control animals, but less of an impact on viral RNA in throat and nasal swabs following SARS-CoV-2 inoculation.
Prophylactic or therapeutic administration of etesevimab to male Rhesus macaques (n=3 per group) resulted in approximately 4 or 3 log10 average decreases, respectively, in viral genomic RNA in oropharyngeal swabs at Day 4 post infection relative to control animals.
The applicability of these findings to a prophylaxis or treatment setting is not known.
18 CLINICAL TRIAL RESULTS AND SUPPORTING DATA FOR EUA
18.1 Treatment of Mild to Moderate COVID-19 (BLAZE-1)
Adults (≥18 Years) and Pediatric Patients (12 to <18 Years Weighing at Least 40 kg)
The data supporting this EUA for treatment of mild to moderate COVID-19 are primarily based on analyses of data from the Phase 2/3 BLAZE-1 trial (NCT04427501). This trial provides Phase 3 placebo-controlled clinical efficacy data from subjects receiving 700 mg bamlanivimab and 1,400 mg of etesevimab together, as well as for subjects receiving 2,800 mg bamlanivimab and 2,800 mg etesevimab together.
BLAZE-1 is a randomized, double-blind, placebo-controlled clinical trial studying bamlanivimab and etesevimab administered together for the treatment of subjects with mild to moderate COVID-19 (subjects with COVID-19 symptoms who are not hospitalized). BLAZE-1 enrolled adult subjects who were not hospitalized and had at least 1 or more COVID-19 symptoms that were at least mild in severity. Treatment was initiated within 3 days of obtaining the clinical sample for the first positive SARS-CoV-2 viral infection determination. Subjects in the Phase 3 portion of the trial met the criteria for high-risk (as defined in Section 2).
Phase 3 Data from BLAZE-1 (bamlanivimab 700 mg and etesevimab 1,400 mg)
In this portion of the trial, subjects were treated with a single infusion of bamlanivimab 700 mg and etesevimab 1,400 mg (N=511) or placebo (N=258). The majority (99.2%) of the patients enrolled in these dose arms met the criteria for high-risk adults (≥18 years of age) that included at least one of the following: age ≥65 years, BMI ≥35, chronic kidney disease, diabetes, immunosuppressive disease, immunosuppressant treatment, or age ≥55 years with cardiovascular disease, hypertension, chronic pulmonary disease or other chronic respiratory disease. Participants ages 12-17 were also enrolled in the trial (10 [2.0%] were treated with bamlanivimab and etesevimab and 13 [1.7%] were treated with placebo), and met high-risk criteria as defined in the trial protocol.
At baseline, median age was 56 years (with 30% of subjects aged 65 or older); 53% of subjects were female, 87% were White, 27% were Hispanic or Latino, and 8% were Black or African American. Subjects had mild (76%) to moderate (24%) COVID-19; the mean duration of symptoms was 4 days; mean viral load by cycle threshold (CT) was 24.33 at baseline. The baseline demographics and disease characteristics were well balanced across treatment groups.
The primary endpoint was the proportion of subjects with COVID-19 related hospitalization (defined as ≥24 hours of acute care) or death by any cause by Day 29. Events occurred in 15 subjects treated with placebo (6%) as compared to 4 events in subjects treated with bamlanivimab 700 mg and etesevimab 1,400 mg together (0.8%) [p<0.0001], an 87% reduction. There were 4 deaths in subjects treated with placebo and no deaths in subjects treated with bamlanivimab 700 mg and etesevimab 1,400 mg together (p=0.01).
Secondary endpoints include mean change in viral load from baseline to Day 3, 5, and 7 (Figure 1).
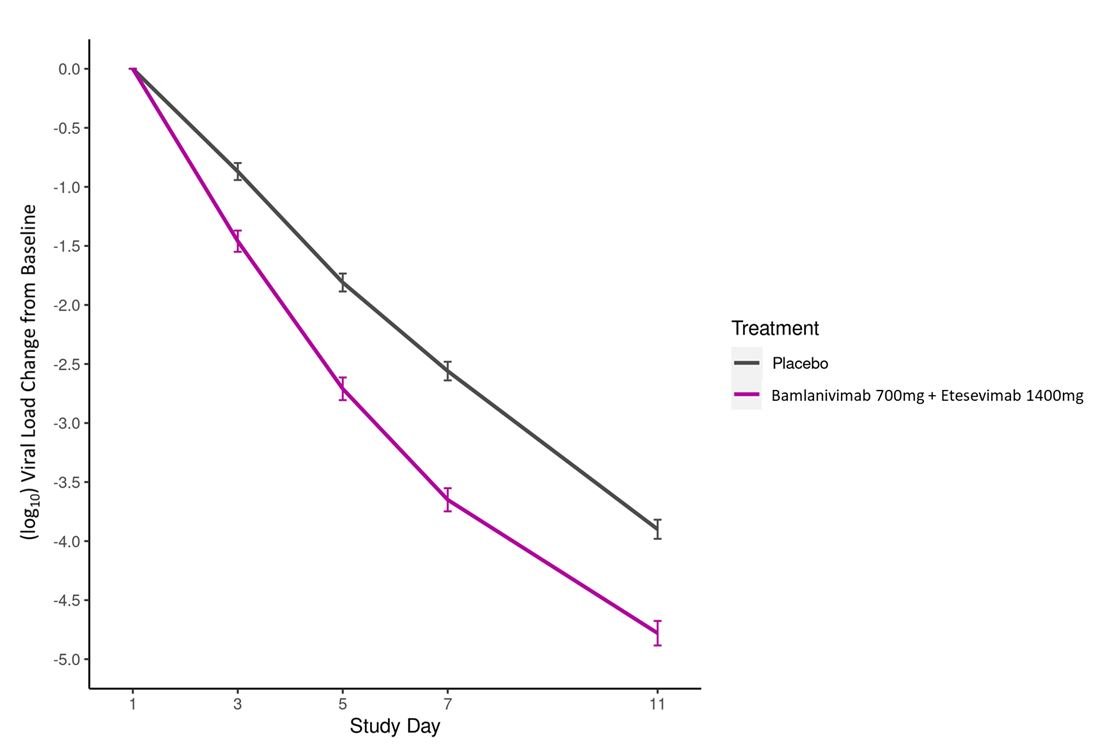
Figure 1: SARS-CoV-2 Viral Load Change from Baseline (Mean ± SE) by Visit from the Phase 3 Portion of BLAZE-1 (700 mg bamlanivimab and 1,400 mg etesevimab).
The median time to sustained symptom resolution as recorded in a trial specific daily symptom diary was 8 days for subjects treated with bamlanivimab 700 mg and etesevimab 1,400 mg together as compared with 10 days for subjects treated with placebo (p=0.009). Symptoms assessed were cough, shortness of breath, feeling feverish, fatigue, body aches and pains, sore throat, chills, and headache. Sustained symptom resolution was defined as absence of any of these symptoms, except for allowance of mild fatigue and cough, in two consecutive assessments.
Phase 3 Data from BLAZE-1 (bamlanivimab 2,800 mg and etesevimab 2,800 mg)
Subjects were treated with a single infusion of bamlanivimab 2,800 mg and etesevimab 2,800 mg (N=518) or placebo (N=517). All of the patients enrolled in these dose arms met the criteria for high-risk adults (≥18 years of age) that included at least one of the following: age ≥65 years of age, BMI ≥35, chronic kidney disease, diabetes, immunosuppressive disease, immunosuppressant treatment, or age ≥55 years with cardiovascular disease, hypertension, chronic pulmonary disease or other chronic respiratory disease. Participants ages 12-17 years were also enrolled in the trial (4 [0.8%] were treated with bamlanivimab and etesevimab and 7 [1.4%] were treated with placebo), and met high-risk criteria as defined in the trial protocol.
Bamlanivimab 2,800 mg and etesevimab 2,800 mg is not an authorized dosage under this EUA. The baseline demographics and disease characteristics were well balanced across treatment groups.
The primary endpoint was the proportion of subjects with COVID-19 related hospitalization (defined as ≥24 hours of acute care) or death by any cause by Day 29. Events occurred in 36 subjects treated with placebo (7%) as compared to 11 events in subjects treated with bamlanivimab 2,800 mg and etesevimab 2,800 mg together (2%) [p<0.001], a 70% reduction. There were 10 deaths in subjects treated with placebo and no deaths in subjects treated with bamlanivimab 2,800 mg and etesevimab 2,800 mg together (p<0.001).
Pediatric Patients <18 Years
The safety and efficacy of bamlanivimab and etesevimab together was evaluated in a total of 125 pediatric subjects enrolled in the Phase 2/3 BLAZE-1 trial (NCT04427501), in which subjects were treated for mild to moderate COVID-19. Pediatric subjects were not hospitalized, and treatment was initiated within 3 days of obtaining the clinical sample for the first positive SARS-CoV-2 viral infection determination. All pediatric subjects met the criteria for high-risk (as defined in Section 2). Pediatric patients weighing 40 kg or more received the same dose as adults (700 mg bamlanivimab and 1,400 mg etesevimab). Pediatric subjects weighing less than 40 kg received weight-based dosing.
Of the 125 pediatric subjects, 33 subjects ages 12 to <18 were evaluated in double-blind, placebo-controlled Phase 3 cohorts of BLAZE-1, and 1 subject age 12 to <18 was evaluated in a controlled addendum to BLAZE-1. Of the 33 pediatric subjects, 14 received placebo, 14 received the authorized dose or a higher dose for their age, and 5 received a lower dose than authorized for their age. A total of 91 pediatric subjects were evaluated in an open-label addendum to BLAZE-1, with 40 subjects ages 12 to <18, 36 ages 6 to <12, 10 ages 2 to <6, and 5 ages 0 to <2. The youngest participant in the trial was 10 months of age and weighed 8.6 kg.
At baseline, median age was 12 years; 46% of subjects were female, 38% were White, 20% were Hispanic or Latino, and 57% were Black or African American. Subjects had mild (88%) to moderate (12%) COVID-19; the mean duration of symptoms was 4 days; mean viral load by cycle threshold (CT) was 5.92 at baseline.
No pediatric subjects died or required hospitalization due to COVID-19. The change in viral load to Day 7 by dose was: -4.23 for subjects treated with 700 mg bamlanivimab and 1,400 mg etesevimab (n=9) and -4.23 for subjects receiving weight-based dosing with bamlanivimab and etesevimab (n=75).
The median time to complete symptom resolution as recorded in a trial specific daily symptom diary was 7 days for subjects treated with bamlanivimab 700 mg and etesevimab 1,400 mg (n=10) and 5 days for subjects treated with weight-based dosing of bamlanivimab and etesevimab (n=91). Symptoms assessed were shortness of breath, nasal congestion, fever, chills, sore throat, stomachache, nausea, vomiting, diarrhea, cough, tiredness, muscle or body aches, headache, new loss of smell, new loss of taste, and poor appetite or poor feeding. Complete symptom resolution was defined as absence of all symptoms at a single timepoint.
18.2 Post-Exposure Prophylaxis of COVID-19 (BLAZE-2)
The data supporting this EUA for post-exposure prophylaxis of COVID-19 are based on the final analysis of Part 1 of the Phase 3 trial BLAZE-2 (NCT04497987). The database lock occurred after all enrolled subjects completed Day 57. BLAZE-2 Part 1 is a randomized, double-blind, placebo-controlled study evaluating bamlanivimab alone for prevention of COVID-19 in residents and staff of skilled nursing facilities following a confirmed reported case of SARS-CoV-2 infection at the facility. No pediatric participants were enrolled.
All participants in Part 1 were randomized and treated with a single infusion of bamlanivimab 4,200 mg or placebo. Results of baseline testing for SARS-CoV-2 were not known until after the therapy was administered. Those with a positive baseline SARS-CoV-2 RT-PCR test were included in the Treatment Population (N=132) and those with a negative test were included in the Prevention Population (N=966). Individuals in these populations were also required to have a baseline negative SARS-CoV-2 serology test; those who tested positive were only included in the overall safety population.
Data are presented for the Prevention Population only. No data were collected on the type or extent of exposure to the index case in the Prevention Population.
In the overall Prevention Population (N=484 for bamlanivimab 4,200 mg and N=482 for placebo) at baseline, the median age was 53 years (with 29% of subjects aged 65 or older); 75% of subjects were female, 89% were White, 5% were Hispanic or Latino, and 8% were Black. The baseline demographics and disease characteristics were well balanced across bamlanivimab and placebo treatment groups.
The primary endpoint (cases of symptomatic COVID-19 by Day 57) was assessed after all participants in the Prevention Population reached 8 weeks of follow-up, and analysis were adjusted for facility, sex, and role within facility (resident/staff). There were 114 cases of symptomatic COVID-19, with a lower frequency occurring in participants treated with bamlanivimab as compared to placebo (residents and staff; adjusted odds ratio 0.43; p<0.001) reducing the risk of being infected with COVID-19 by up to 57%. As a supplementary analysis, the time to symptomatic COVID-19 is shown for each arm in Figure 2. Four COVID-19-related deaths were reported in the overall Prevention Population; all occurred in the placebo arm (0.8%). No COVID-19-related deaths occurred in the bamlanivimab arm.
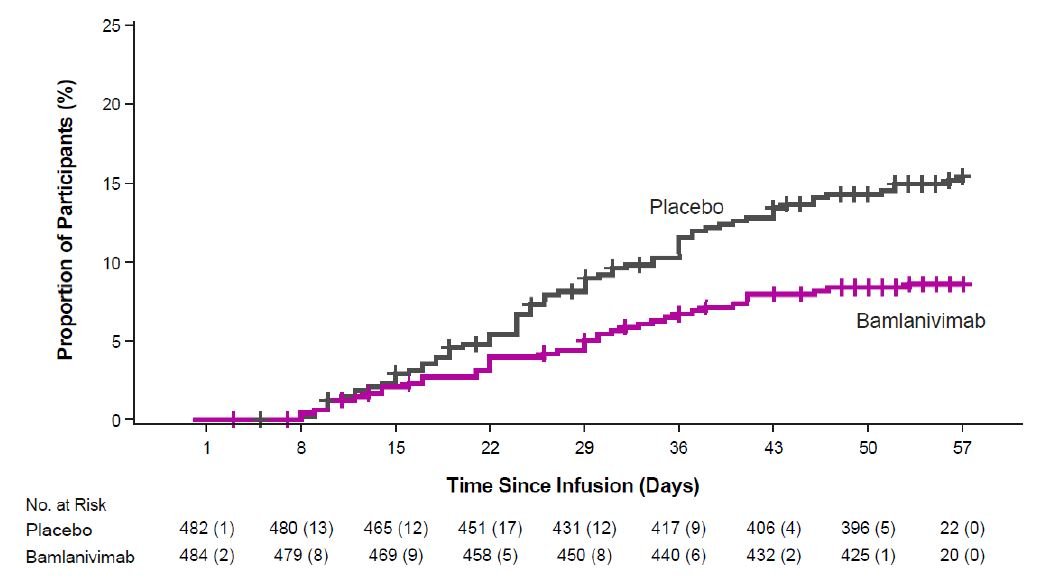
Figure 2: Time to symptomatic COVID-19 in the overall prevention population (residents and staff).
For the pre-specified subgroup of nursing home residents, there were 45 cases of symptomatic COVID-19, with a lower frequency in those treated with bamlanivimab versus placebo (adjusted odds ratio 0.20; p<0.001), reducing the risk of being infected with COVID-19 by up to 80%. The time to symptomatic COVID-19 in nursing home residents is shown by treatment arm in Figure 3. In this same cohort of residents within the Prevention Population, 6 deaths due to any cause occurred in residents treated with placebo (4.3%) and 5 deaths due to any cause occurred in residents treated with bamlanivimab (3.1%).
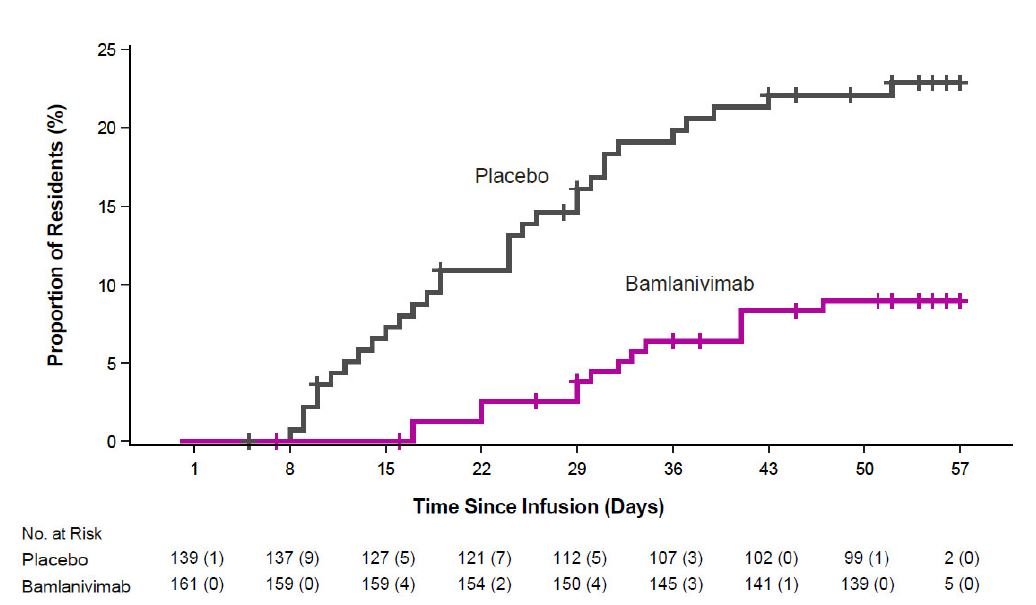
Figure 3: Time to symptomatic COVID-19 in residents only.
For the post-hoc subgroup of patients who met the high risk criteria (all residents and all high risk staff7), there were 75 cases of symptomatic COVID-19, with a lower frequency in those treated with bamlanivimab versus placebo (adjusted odds ratio 0.28; nominal p<0.001), reducing the risk of being infected with COVID-19 by up to 72%.
For the post-hoc subgroup of staff who did not meet high risk criteria, there were 39 cases of symptomatic COVID-19, with less evidence of a preventative effect for bamlanivimab versus placebo (adjusted odds ratio 0.64; nominal p=0.26).
- 7
- All high risk participants in the Prevention Population were either residents in a skilled nursing or assisted living facility, or staff in a skilled nursing or assisted living facility who satisfied at least 1 of the following at the time of screening: were ≥65 years of age, had a BMI ≥35, had CKD, had diabetes, had immunosuppressive disease, were currently receiving immunosuppressive treatment, OR were ≥55 years of age AND had cardiovascular disease, OR hypertension, OR COPD or other chronic respiratory disease.
19. How is Bamlanivimab and Etesevimab Injection supplied
How Supplied
UNDER THIS EUA, BAMLANIVIMAB AND ETESEVIMAB MUST BE ADMINISTERED TOGETHER.
Bamlanivimab
Bamlanivimab injection is a sterile, preservative-free clear to opalescent and colorless to slightly yellow to slightly brown solution supplied in a vial.
Etesevimab
Etesevimab injection is a sterile, preservative-free clear to opalescent and colorless to slightly yellow to slightly brown solution supplied in a vial.
Bamlanivimab and etesevimab are supplied as:
| Antibody | Concentration | Package Size | NDC |
| Bamlanivimab | 700 mg/20 mL (35 mg/mL) | one vial per carton |
0002-7910-01 |
| Etesevimab | 700 mg/20 mL (35 mg/mL) | one vial per carton |
0002-7950-01 |
Storage and Handling
Bamlanivimab is preservative-free. Discard unused portion.
Etesevimab is preservative-free. Discard unused portion.
Store unopened vials in a refrigerator at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light.
FDA has authorized an extension to the shelf-life (i.e., expiration date) of both bamlanivimab and etesevimab following a thorough review of data submitted by Eli Lilly and Company. The extension applies to all unopened vials of bamlanivimab and etesevimab that have been held in accordance with storage conditions. Confirm the shelf-life of unopened vials of bamlanivimab and etesevimab by batch number at the FDA EUA website under the Drug and Biological Therapeutic Products bamlanivimab and etesevimab. This site includes a complete listing of extended expiration dates by batch number. If the batch number on the vial/carton is not included in this listing, the product is labeled with the correct expiration date.
DO NOT FREEZE, SHAKE, OR EXPOSE TO DIRECT LIGHT.
The prepared infusion solution is intended to be used immediately. If immediate administration is not possible, store infusion solution in the refrigerator at 2°C to 8°C (36°F to 46°F) for up to 24 hours and at room temperature (20°C to 25°C [68°F to 77°F]) and for up to 7 hours, including infusion time. If refrigerated, allow the infusion solution to equilibrate to room temperature prior to administration.
20. Patient Counseling Information
Patients treated with bamlanivimab and etesevimab should continue to self-isolate and use infection control measures (e.g., wear mask, isolate, social distance, avoid sharing personal items, clean and disinfect “high touch” surfaces, and frequent handwashing) according to CDC guidelines. Also see Fact Sheet for Patients, Parents and Caregivers.
21 CONTACT INFORMATION
For additional information visit:
www.LillyAntibody.com
If you have questions, please contact:
1-855-LillyC19 (1-855-545-5921)
Literature revised January 24, 2022
Eli Lilly and Company, Indianapolis, IN 46285, USA
Copyright © 2021, 2022, Eli Lilly and Company. All rights reserved.
ETE-0009-EUA HCP-20220124
Fact Sheet for Patients, Parents and Caregivers
Emergency Use Authorization (EUA) of Bamlanivimab and Etesevimab for Coronavirus Disease 2019 (COVID-19)
You or your child are being given two medicines together called bamlanivimab and etesevimab for the treatment or post-exposure prophylaxis for prevention of coronavirus disease 2019 (COVID-19). SARS-CoV-2 is the virus that causes COVID-19. This Fact Sheet contains information to help you understand the potential risks and potential benefits of taking bamlanivimab and etesevimab.
Receiving bamlanivimab and etesevimab may help to treat COVID-19 in certain people, or help to prevent COVID-19 in certain people who have been exposed to someone infected with SARS-CoV-2 or who are at high risk of an exposure because of being in the same setting, such as nursing homes or prisons.
Read this Fact Sheet for information about bamlanivimab and etesevimab. Talk to your or your child’s healthcare provider if you have questions. It is your choice if you or your child receive bamlanivimab and etesevimab or you may stop them at any time.
What is COVID-19?
COVID-19 is caused by a virus called a coronavirus, SARS-CoV-2. People can get COVID-19 through contact with another person who has the virus.
COVID-19 illnesses have ranged from very mild (including some with no reported symptoms) to severe, including illness resulting in death. While information so far suggests that most COVID-19 illness is mild, serious illness can happen and may cause some of your or your child’s other medical conditions to become worse. People of all ages with severe, long-lasting (chronic) medical conditions like heart disease, lung disease, and diabetes, for example, and other conditions including obesity, seem to be at higher risk of being hospitalized for COVID-19. Older age, with or without other conditions, also places people at higher risk of being hospitalized for COVID-19.
What are the symptoms of COVID-19?
The symptoms of COVID-19 include fever, cough, and shortness of breath, which may appear 2 to 14 days after exposure. Serious illness including breathing problems can occur and may cause other medical conditions to become worse.
What are bamlanivimab and etesevimab?
Bamlanivimab and etesevimab are investigational medicines used together in adults and children who are at high risk for developing severe COVID-19, including hospitalization or death for:
- treatment of mild to moderate symptoms of COVID-19, OR
-
post-exposure prophylaxis for prevention of COVID-19 in persons who are:
- not fully vaccinated against COVID-19 (Individuals are considered to be fully vaccinated 2 weeks after their second dose in a 2-dose series [such as the Pfizer or Moderna vaccines], or 2 weeks after a single-dose dose vaccine [such as Johnson & Johnson’s Janssen vaccine]), or
- are not expected to build up enough of an immune response to the complete COVID-19 vaccination (for example, someone with immunocompromising conditions, including someone who is taking immunosuppressive medications), and
- have been exposed to someone who is infected with SARS-CoV-2. Close contact with someone who is infected with SARS-CoV-2 is defined as being within 6 feet for a total of 15 minutes or more, providing care at home to someone who is sick, having direct physical contact with the person (hugging or kissing, for example), sharing eating or drinking utensils, or being exposed to respiratory droplets from an infected person (sneezing or coughing, for example). For additional details, go to https://www.cdc.gov/coronavirus/2019-ncov/if-you-are-sick/quarantine.html, or
- someone who is at high risk of being exposed to someone who is infected with SARS-CoV-2 because of occurrence of SARS-CoV-2 infection in other individuals in the same institutional setting (for example, nursing homes, prisons).
Bamlanivimab and etesevimab are investigational because they are still being studied. There is limited information known about the safety or effectiveness of using bamlanivimab and etesevimab to treatment or prevention of COVID-19. Bamlanivimab and etesevimab are not authorized for pre-exposure prophylaxis for prevention of COVID-19.
The FDA has authorized the emergency use of bamlanivimab and etesevimab together for the treatment of COVID-19 and the post-exposure prophylaxis for prevention of COVID-19 under an Emergency Use Authorization (EUA). For more information on EUA, see the section “What is an Emergency Use Authorization (EUA)?” at the end of this Fact Sheet.
What should I tell the healthcare provider before I or my child receive bamlanivimab and etesevimab?
Tell the healthcare provider about all of your or your child’s medical conditions, including:
- Having any allergies
- Having received a COVID-19 vaccine
- Having any serious illnesses
- Are pregnant or plan to become pregnant
- Are breastfeeding or plan to breastfeed
- Are taking any medications (prescription, over-the-counter, vitamins, and herbal products)
How are bamlanivimab and etesevimab given?
- Bamlanivimab and etesevimab are given at the same time through a vein (intravenous or IV).
- One dose of bamlanivimab and etesevimab will be given by IV infusion. The infusion will take 16 – 60 minutes or longer. Your or your child’s healthcare provider will determine the duration of the infusion.
What are the important possible side effects of bamlanivimab and etesevimab?
Possible side effects of bamlanivimab and etesevimab are:
- Allergic reactions. Allergic reactions can happen during and after infusion with bamlanivimab and etesevimab. Tell your or your child’s healthcare provider right away if any of the following signs and symptoms of allergic reactions occur: fever, chills, nausea, headache, shortness of breath, low or high blood pressure, rapid or slow heart rate, chest discomfort or pain, weakness, confusion, feeling tired, wheezing, swelling of the lips, face, or throat, rash including hives, itching, muscle aches, feeling faint, dizziness, and sweating. These reactions may be severe or life threatening.
- Worsening of COVID-19 symptoms after bamlanivimab and etesevimab therapy for active infection: You or your child may experience new or worsening symptoms after infusion for mild to moderate COVID-19, including fever, difficulty breathing, rapid or slow heart rate, tiredness, weakness or confusion. If these occur, contact your or your child’s healthcare provider or seek immediate medical attention as some of these events have required hospitalization. It is unknown if these events are related to treatment or are due to the progression of COVID-19.
The side effects of getting any medicine by vein may include brief pain, bleeding, bruising of the skin, soreness, swelling, and possible infection at the infusion site.
These are not all the possible side effects of bamlanivimab and etesevimab. Not a lot of people have been given bamlanivimab and etesevimab. Serious and unexpected side effects may happen. Bamlanivimab and etesevimab are still being studied so it is possible that all of the risks are not known at this time.
It is possible that bamlanivimab and etesevimab could interfere with your or your child’s body’s own ability to fight off a future infection of SARS-CoV-2. Similarly, bamlanivimab and etesevimab may reduce the body’s immune response to a vaccine for SARS-CoV-2. Specific studies have not been conducted to address these possible risks. Talk to your or your child’s healthcare provider if you have any questions.
What other treatment choices are there?
Like bamlanivimab and etesevimab, FDA may allow for the emergency use of other medicines to treat people with COVID-19. Go to https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization for information on the emergency use of other medicines that are not approved by FDA to treat people with COVID-19. Your or your child’s healthcare provider may talk with you about clinical trials you or your child may be eligible for.
It is your choice whether you or your child should be treated or not to be treated with bamlanivimab and etesevimab. Should you decide that you or your child should not receive bamlanivimab and etesevimab or stop it at any time, it will not change your or your child’s standard medical care.
What other prevention choices are there?
Vaccines to prevent COVID-19 are approved or available under Emergency Use Authorization. Use of bamlanivimab and etesevimab does not replace vaccination against COVID-19.
Like bamlanivimab and etesevimab, FDA may allow for the emergency use of other medicines for post-exposure prophylaxis for prevention of COVID-19. Go to https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization for information on the emergency use of other medicines that are not approved by FDA for post-exposure prophylaxis for prevention of COVID-19. The healthcare provider may talk with you about clinical trials you or your child may be eligible for.
Bamlanivimab and etesevimab are not authorized for pre-exposure prophylaxis for prevention of COVID-19.
What if I am pregnant or breastfeeding?
There is limited experience treating pregnant women or breastfeeding mothers with bamlanivimab and etesevimab. For a mother and unborn baby, the benefit of receiving bamlanivimab and etesevimab may be greater than the risk from the treatment. If pregnant or breastfeeding, discuss your options and specific situation with your healthcare provider.
How do I report side effects with bamlanivimab and etesevimab?
Tell the healthcare provider right away if you or your child have any side effect that bothers you or your child, or does not go away.
Report side effects to FDA MedWatch at www.fda.gov/medwatch, call 1-800-FDA-1088, or contact Eli Lilly and Company at 1-855-LillyC19 (1-855-545-5921).
How can I learn more?
- Ask your or your child’s healthcare provider
- Visit www.LillyAntibody.com
- Visit https://www.covid19treatmentguidelines.nih.gov/
- Contact your local or state public health department
What is an Emergency Use Authorization (EUA)?
The United States FDA has made bamlanivimab and etesevimab available under an emergency access mechanism called an EUA. The EUA is supported by a Secretary of Health and Human Service (HHS) declaration that circumstances exist to justify the emergency use of drugs and biological products during the COVID-19 pandemic.
Bamlanivimab and etesevimab have not undergone the same type of review as an FDA-approved product. In issuing an EUA under the COVID-19 public health emergency, the FDA must determine, among other things, that based on the totality of scientific evidence available, it is reasonable to believe that the product may be effective for diagnosing, treating, or preventing COVID-19, or a serious or life-threatening disease or condition caused by COVID-19; that the known and potential benefits of the product, when used to diagnose, treat, or prevent such disease or condition, outweigh the known and potential risks of such product; and that there are no adequate, approved and available alternatives. All of these criteria must be met to allow for the medicine to be used in the treatment of COVID-19 or prevention of COVID-19 during the COVID-19 pandemic.
The EUA for bamlanivimab and etesevimab together is in effect for the duration of the COVID-19 declaration justifying emergency use of these products, unless terminated or revoked (after which the products may no longer be used).
Literature revised December 3, 2021
Eli Lilly and Company, Indianapolis, IN 46285, USA
ETE-0005-EUA PAT-20211203
PACKAGE LABEL- etesevimab Injection 700 mg/20 mL (35 mg/mL) Vial Carton
NDC 0002-7950-01
Lilly
etesevimab injection
700 mg/20 mL
(35 mg/mL)
For Intravenous Infusion Only
Must dilute before use
Single-Dose Vial: Discard Unused Portion
For use under Emergency Use Authorization (EUA).
MUST ADMINISTER WITH BAMLANIVIMAB

Copyright © 2021, Eli Lilly and Company. All rights reserved.
RECENT MAJOR CHANGES
|
Revised 01/2022 |
|
Revised 12/2021 |
|
Revised 12/2021, 08/2021, 05/2021, and 03/2021 |
|
Revised 12/2021 |
|
Revised 12/2021 |
|
Revised 09/2021 |
|
Revised 09/2021 |
|
Revised 08/2021 |
|
Revised 08/2021 |
|
Revised 08/2021 |
|
Revised 08/2021 |
|
Revised 05/2021 |
|
Revised 05/2021 |
|
Revised 05/2021 |
|
Revised 05/2021 |
Bamlanivimab and etesevimab have been authorized by FDA for the emergency uses described above.
Bamlanivimab and etesevimab are not FDA-approved for these uses.
Bamlanivimab and etesevimab are authorized only for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of bamlanivimab and etesevimab under section 564(b)(1) of the Act, 21 U.S.C. § 360bbb-3(b)(1), unless the authorization is terminated or revoked sooner.
Dosing
See Full Fact Sheet for Healthcare Providers for information on dosing [see Dosage and Administration (2)].
Preparation and Administration
See Full Fact Sheet for Healthcare Providers for information on preparation and administration [see Dose Preparation and Administration (2.4)].
Under this EUA, single-dose vials may be used to prepare more than one pediatric dose; in addition, pediatric doses do not need to be diluted for patients <18 years and weighing <40kg.
Storage and Handling
Refrigerate unopened vials at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light. Do not freeze, shake, or expose to direct light.
FDA has authorized an extension to the shelf-life (i.e., expiration date) of both bamlanivimab and etesevimab following a thorough review of data submitted by Eli Lilly and Company. The extension applies to all unopened vials of bamlanivimab and etesevimab that have been held in accordance with storage conditions. Confirm the shelf-life of unopened vials of bamlanivimab and etesevimab by batch number at the FDA EUA website under the Drug and Biological Therapeutic Products bamlanivimab and etesevimab. This site includes a complete listing of extended expiration dates by batch number. If the batch number on the vial/carton is not included in this listing, the product is labeled with the correct expiration date.
Warnings
There are limited clinical data available for bamlanivimab and etesevimab. Serious and unexpected adverse events may occur that have not been previously reported with use of bamlanivimab and etesevimab together.
Hypersensitivity Including Anaphylaxis and Infusion-Related Reactions
Serious hypersensitivity reactions, including anaphylaxis, have been observed with administration of bamlanivimab and etesevimab. If signs and symptoms of a clinically significant hypersensitivity reaction or anaphylaxis occur, immediately discontinue administration and initiate appropriate medications and/or supportive therapy.
Infusion-related reactions, occurring during the infusion and up to 24 hours after the infusion, have been observed with administration of bamlanivimab and etesevimab together. These reactions may be severe or life threatening.
Signs and symptoms of infusion related reactions may include:
- fever, difficulty breathing, reduced oxygen saturation, chills, fatigue, arrhythmia (e.g., atrial fibrillation, sinus tachycardia, bradycardia), chest pain or discomfort, weakness, altered mental status, nausea, headache, bronchospasm, hypotension, hypertension, angioedema, throat irritation, rash including urticaria, pruritus, myalgia, vasovagal reactions (e.g., pre-syncope, syncope), dizziness and diaphoresis.
Consider slowing or stopping the infusion and administer appropriate medications and/or supportive care if an infusion-related reaction occurs.
Hypersensitivity reactions occurring more than 24 hours after the infusion have also been reported with the use of bamlanivimab and etesevimab under Emergency Use Authorization.
Clinical Worsening After Bamlanivimab and Etesevimab Administration
Clinical worsening of COVID-19 after administration of bamlanivimab and etesevimab together has been reported and may include signs or symptoms of fever, hypoxia or increased respiratory difficulty, arrhythmia (e.g., atrial fibrillation, sinus tachycardia, bradycardia), fatigue, and altered mental status. Some of these events required hospitalization. It is not known if these events were related to bamlanivimab and etesevimab use or were due to progression of COVID-19.
Limitations of Benefit and Potential for Risk in Patients with Severe COVID-19
Treatment with bamlanivimab and etesevimab has not been studied in patients hospitalized due to COVID-19. Monoclonal antibodies, such as bamlanivimab and etesevimab, may be associated with worse clinical outcomes when administered to hospitalized patients with COVID-19 requiring high flow oxygen or mechanical ventilation. Therefore, [see Limitations of Authorized Use (1.1)]:
- Bamlanivimab and etesevimab are not authorized for use in patients 2 years and older who are hospitalized due to COVID-192,
- Bamlanivimab and etesevimab are not authorized for use in patients, regardless of age, who:
- require oxygen therapy and/or respiratory support due to COVID-19, OR
- require an increase in baseline oxygen flow rate and/or respiratory support due to COVID-19 and are on chronic oxygen therapy and/or respiratory support due to underlying non-COVID-19 related comorbidity.
Side Effects
Adverse events have been reported with bamlanivimab and etesevimab [see Full EUA Prescribing Information, Overall Safety Summary (6.1)].
Additional adverse events associated with bamlanivimab and etesevimab, some of which may be serious, may become apparent with more widespread use.
INSTRUCTIONS FOR HEALTHCARE PROVIDERS
As the healthcare provider, you must communicate to your patient or parent/caregiver, as age appropriate, information consistent with the “Fact Sheet for Patients, Parents and Caregivers” (and provide a copy of the Fact Sheet) prior to the patient receiving bamlanivimab and etesevimab, including:
- FDA has authorized the emergency use of bamlanivimab and etesevimab administered together for the treatment of mild to moderate COVID-19 in adults and pediatric patients, including neonates, with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death [see Limitations of Authorized Use (1.1)].
- FDA has authorized the emergency use of bamlanivimab and etesevimab administered together in adults and pediatric individuals, including neonates, for post-exposure prophylaxis of COVID-19 in individuals who are at high risk for progression to severe COVID-19, including hospitalization or death, and are:
- not fully vaccinated3 or who are not expected to mount an adequate immune response to complete SARS-CoV-2 vaccination (for example, individuals with immunocompromising conditions including those taking immunosuppressive medications4) and
- have been exposed to an individual infected with SARS-CoV-2 consistent with close contact criteria per Centers for Disease Control and Prevention (CDC)5 or
- who are at high risk of exposure to an individual infected with SARS-CoV-2 because of occurrence of SARS-CoV-2 infection in other individuals in the same institutional setting (for example, nursing homes, prisons) [see Limitations of Authorized Use (1.2)].
- not fully vaccinated3 or who are not expected to mount an adequate immune response to complete SARS-CoV-2 vaccination (for example, individuals with immunocompromising conditions including those taking immunosuppressive medications4) and
- The patient or parent/caregiver has the option to accept or refuse bamlanivimab and etesevimab.
- The significant known and potential risks and benefits of bamlanivimab and etesevimab, and the extent to which such potential risks and benefits are unknown.
- Information on available alternative treatments and the risks and benefits of those alternatives, including clinical trials.
- Patients treated with bamlanivimab and etesevimab together should continue to self-isolate and use infection control measures (e.g., wear mask, isolate, social distance, avoid sharing personal items, clean and disinfect “high touch” surfaces, and frequent handwashing) according to CDC guidelines.
For information on clinical trials that are testing the use of bamlanivimab and etesevimab together for COVID-19, please see www.clinicaltrials.gov.
MANDATORY REQUIREMENTS FOR BAMLANIVIMAB AND ETESEVIMAB ADMINISTRATION UNDER EMERGENCY USE AUTHORIZATION:
In order to mitigate the risks of using these unapproved products and to optimize the potential benefit of bamlanivimab and etesevimab under this EUA, the following items are required. Use of bamlanivimab and etesevimab under this EUA is limited to the following (all requirements must be met):
- Treatment of mild to moderate COVID-19 in adults and pediatric patients, including neonates, with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death [see Limitations of Authorized Use (1.1)].
- Post-exposure prophylaxis of COVID-19 in adults and pediatric individuals, including neonates, who are at high risk for progression to severe COVID-19, including hospitalization or death, and are:
- not fully vaccinated3 or who are not expected to mount an adequate immune response to complete SARS-CoV-2 vaccination (for example, individuals with immunocompromising conditions including those taking immunosuppressive medications4) and
- have been exposed to an individual infected with SARS-CoV-2 consistent with close contact criteria per Centers for Disease Control and Prevention (CDC)5 or
- who are at high risk of exposure to an individual infected with SARS-CoV-2 because of occurrence of SARS-CoV-2 infection in other individuals in the same institutional setting (for example, nursing homes, prisons) [see Limitations of Authorized Use (1.2)].
- not fully vaccinated3 or who are not expected to mount an adequate immune response to complete SARS-CoV-2 vaccination (for example, individuals with immunocompromising conditions including those taking immunosuppressive medications4) and
- As the healthcare provider, communicate to your patient or parent/caregiver, as age appropriate, information consistent with the “Fact Sheet for Patients, Parents and Caregivers” prior to the patient receiving bamlanivimab and etesevimab. Healthcare providers (to the extent practicable given the circumstances of the emergency) must document in the patient’s medical record that the patient/caregiver has been:
- Given the “Fact Sheet for Patients, Parents and Caregivers”,
- Informed of alternatives to receiving authorized bamlanivimab and etesevimab, and
- Informed that bamlanivimab and etesevimab are unapproved drugs that are authorized for use under this Emergency Use Authorization.
- Patients with known hypersensitivity to any ingredient of bamlanivimab or etesevimab must not receive bamlanivimab and etesevimab.
- The prescribing health care provider and/or the provider’s designee is/are responsible for mandatory reporting of all medication errors and serious adverse events* potentially related to bamlanivimab and etesevimab treatment within 7 calendar days from the onset of the event. The reports must include unique identifiers and the words “bamlanivimab and etesevimab use for COVID-19 under Emergency Use Authorization (EUA)” in the description section of the report.
- Submit adverse event reports to FDA MedWatch using one of the following methods:
- Complete and submit the report online: www.fda.gov/medwatch/report.htm, or
- Complete and submit a postage-paid FDA Form 3500 (https://www.fda.gov/media/76299/download) and return by:
- Mail to MedWatch, 5600 Fishers Lane, Rockville, MD 20852-9787, or
- Fax (1-800-FDA-0178), or
- Call 1-800-FDA-1088 to request a reporting form.
- Submitted reports must include in the field name, “Describe Event, Problem, or Product Use/Medication Error” the statement “bamlanivimab and etesevimab use for COVID-19 under Emergency Use Authorization (EUA).”
*Serious Adverse Events are defined as:
- death;
- a life-threatening adverse event;
- inpatient hospitalization or prolongation of existing hospitalization;
- a persistent or significant incapacity or substantial disruption of the ability to conduct normal life functions;
- a congenital anomaly/birth defect;
- a medical or surgical intervention to prevent death, a life-threatening event, hospitalization, disability, or congenital anomaly.
- Submit adverse event reports to FDA MedWatch using one of the following methods:
- The prescribing health care provider and/or the provider’s designee is/are to provide mandatory responses to requests from FDA for information about adverse events and medication errors following receipt of bamlanivimab and etesevimab.
- OTHER REPORTING REQUIREMENTS
- Healthcare facilities and providers must report therapeutics information and utilization data through HHS Protect, Teletracking or National Healthcare Safety Network (NHSN) as directed by the U.S. Department of Health and Human Services.
- In addition, please provide a copy of all FDA MedWatch forms to:
Eli Lilly and Company, Global Patient Safety
Fax: 1-317-277-0853
E-mail: mailindata_gsmtindy@lilly.com
Or call Eli Lilly and Company at 1-855-LillyC19 (1-855-545-5921) to report adverse events.
APPROVED AVAILABLE ALTERNATIVES
Veklury (remdesivir) is FDA-approved for the treatment of COVID-19 in adults and pediatric patients (12 years of age and older weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing, who are not hospitalized and have mild-to-moderate COVID-19, and who are at high risk for progression to severe COVID-19, including hospitalization or death. Veklury is administered via intravenous infusion for a total treatment duration of 3 days.
Although Veklury is an approved alternative treatment of mild-to-moderate COVID-19 in adults and pediatric patients (12 years of age and older weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death, FDA does not consider Veklury to be an adequate alternative to bamlanivimab and etesevimab for this authorized use because it may not be feasible or practical for certain patients (e.g., it requires a 3-day treatment duration).6
There is no adequate, approved and available alternative to bamlanivimab and etesevimab administered together for post-exposure prophylaxis of COVID-19 in adult and pediatric individuals, including neonates, who are at high risk for progression to severe COVID-19, including hospitalization or death, and are:
- not fully vaccinated3 or who are not expected to mount an adequate immune response to complete SARS-CoV-2 vaccination (for example, individuals with immunocompromising conditions including those taking immunosuppressive medications4) and
- have been exposed to an individual infected with SARS-CoV-2 consistent with close contact criteria per Centers for Disease Control and Prevention (CDC)5 or
- who are at high risk of exposure to an individual infected with SARS-CoV-2 because of occurrence of SARS-CoV-2 infection in other individuals in the same institutional setting (for example, nursing homes, prisons) [see Limitations of Authorized Use (1.2)].
Additional information on COVID-19 therapies can be found at https://www.cdc.gov/coronavirus/2019-ncov/index.html. The health care provider should visit https://clinicaltrials.gov/ to determine whether the patient may be eligible for enrollment in a clinical trial.
- 6
- Additionally, the approval for Veklury does not cover certain pediatric patients for whom bamlanivimab and etesevimab administered together is authorized (e.g., patients less than 12 years of age).
AUTHORITY FOR ISSUANCE OF THE EUA
The Secretary of the Department of Health and Human Services (HHS) has declared a public health emergency that justifies the emergency use of drugs and biological products during the COVID-19 pandemic. FDA has issued this EUA, requested by Eli Lilly and Company for the unapproved products bamlanivimab and etesevimab administered together for the treatment of mild to moderate COVID-19 in adults and pediatric patients, including neonates, with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death.5
FDA has also issued this EUA, requested by Eli Lilly and Company for the unapproved products bamlanivimab and etesevimab administered together in adults and pediatric individuals, including neonates, for post-exposure prophylaxis of COVID-19 in individuals who are at high risk of progression to severe COVID-19, including hospitalization or death, and are:
- not fully vaccinated3 or who are not expected to mount an adequate immune response to complete SARS-CoV-2 vaccination (for example, individuals with immunocompromising conditions including those taking immunosuppressive medications4) and
- have been exposed to an individual infected with SARS-CoV-2 consistent with close contact criteria per Centers for Disease Control and Prevention (CDC)5 or
- who are at high risk of exposure to an individual infected with SARS-CoV-2 because of occurrence of SARS-CoV-2 infection in other individuals in the same institutional setting (for example, nursing homes, prisons) [see Limitations of Authorized Use (1.2)].
Although limited scientific information is available, based on the totality of the scientific evidence available to date, it is reasonable to believe that bamlanivimab and etesevimab administered together may be effective for the treatment of mild to moderate COVID-19 or for post-exposure prophylaxis of COVID-19 in individuals as specified in this Fact Sheet. You may be contacted and asked to provide information to help with the assessment of the use of the product during this emergency.
This EUA for bamlanivimab and etesevimab will end when the Secretary determines that the circumstances justifying the EUA no longer exist or when there is a change in the approval status of the product such that an EUA is no longer needed.
As a health care provider, you must comply with the mandatory requirements of the EUA (see above).
CONTACT INFORMATION
For additional information visit
www.LillyAntibody.com
If you have questions, please contact
1-855-LillyC19 (1-855-545-5921)
| END SHORT VERSION FACT SHEET Long Version Begins on Next Page |
FULL EUA PRESCRIBING INFORMATION
| FULL EUA PRESCRIBING INFORMATION: CONTENTS* |
11.2 Lactation | ||
| 1 AUTHORIZED USE |
11.3 Pediatric Use | ||
| 1.1 Treatment | 11.4 Geriatric Use | ||
| 1.2 Post-Exposure Prophylaxis | 11.5 Renal Impairment | ||
| 2 DOSAGE AND ADMINISTRATION |
11.6 Hepatic Impairment | ||
| 2.1 Patient Selection | 11.7 Other Specific Populations | ||
| 2.2 Dosage | 12 OVERDOSAGE |
||
| 2.3 Dosage Adjustment in Specific Populations | 13 DESCRIPTION |
||
| 2.4 Dose Preparation and Administration | 14 CLINICAL PHARMACOLOGY |
||
| 3 DOSAGE FORMS AND STRENGTHS |
14.1 Mechanism of Action | ||
| 4 CONTRAINDICATIONS |
14.2 Pharmacodynamics | ||
| 5 WARNINGS AND PRECAUTIONS |
14.3 Pharmacokinetics | ||
| 5.1 Hypersensitivity Including Anaphylaxis and Infusion-Related Reactions | 15 MICROBIOLOGY/RESISTANCE INFORMATION |
||
| 5.2 Clinical Worsening After Bamlanivimab and Etesevimab Administration | 16 NONCLINICAL TOXICOLOGY |
||
| 5.3 Limitations of Benefit and Potential for Risk in Patients with Severe COVID-19 | 17 ANIMAL PHARMACOLOGIC AND EFFICACY DATA |
||
| 6 OVERALL SAFETY SUMMARY |
18 CLINICAL TRIAL RESULTS AND SUPPORTING DATA FOR EUA | ||
| 6.1 Clinical Trials Experience | 18.1 Treatment of Mild to Moderate COVID-19 (BLAZE-1) | ||
| 7 PATIENT MONITORING RECOMMENDATIONS |
18.2 Post-Exposure Prophylaxis of COVID-19 (BLAZE-2) |
||
| 8 ADVERSE REACTIONS AND MEDICATION ERRORS REPORTING REQUIREMENTS AND INSTRUCTIONS |
19 HOW SUPPLIED/STORAGE AND HANDLING |
||
| 9 OTHER REPORTING REQUIREMENTS |
20 PATIENT COUNSELING INFORMATION |
||
| 10 DRUG INTERACTIONS |
21 CONTACT INFORMATION |
||
| 11 USE IN SPECIFIC POPULATIONS |
* Sections or subsections omitted from the full prescribing information are not listed. | ||
| 11.1 Pregnancy |
1 AUTHORIZED USE
1.1 TREATMENT
Bamlanivimab and etesevimab administered together are authorized for use under an EUA for the treatment of mild to moderate coronavirus disease 2019 (COVID-19) in adults and pediatric patients, including neonates, with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death.
Limitations of Authorized Use
- Bamlanivimab and etesevimab are not authorized for treatment of mild to moderate COVID-19 in geographic regions where infection is likely to have been caused by a non-susceptible SARS-CoV-2 variant based on available information including variant susceptibility to these drugs and regional variant frequency.
- FDA’s determination and any updates will be available at: https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization#coviddrugs.1
- Bamlanivimab and etesevimab are not authorized for use in patients 2 years and older who are hospitalized due to COVID-19.2
- Bamlanivimab and etesevimab are not authorized for use in patients, regardless of age, who:
- require oxygen therapy and/or respiratory support due to COVID-19, OR
- require an increase in baseline oxygen flow rate and/or respiratory support due to COVID-19 and are on chronic oxygen therapy and/or respiratory support due to underlying non-COVID-19 related comorbidity.
- Treatment with bamlanivimab and etesevimab has not been studied in patients hospitalized due to COVID-19. Monoclonal antibodies, such as bamlanivimab and etesevimab, may be associated with worse clinical outcomes when administered to hospitalized patients with COVID-19 requiring high flow oxygen or mechanical ventilation [see Warnings and Precautions (5.3)].
1.2 POST-EXPOSURE PROPHYLAXIS
Bamlanivimab and etesevimab administered together are authorized for use under an EUA for post-exposure prophylaxis of COVID-19 in adults and pediatric individuals, including neonates, who are at high risk for progression to severe COVID-19, including hospitalization or death, and are:
- not fully vaccinated3 or who are not expected to mount an adequate immune response to complete SARS-CoV-2 vaccination (for example, individuals with immunocompromising conditions including those taking immunosuppressive medications4) and
- have been exposed to an individual infected with SARS-CoV-2 consistent with close contact criteria per Centers for Disease Control and Prevention (CDC)5 or
- who are at high risk of exposure to an individual infected with SARS-CoV-2 because of occurrence of SARS-CoV-2 infection in other individuals in the same institutional setting (for example, nursing homes, prisons).
Limitations of Authorized Use
- Bamlanivimab and etesevimab are not authorized for post-exposure prophylaxis of COVID-19 in geographic regions where exposure is likely to have been to a non-susceptible SARS-CoV-2 variant based on available information including variant susceptibility to these drugs and regional variant frequency.
- FDA’s determination and any updates will be available at: https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization#coviddrugs.1
- Post-exposure prophylaxis with bamlanivimab and etesevimab is not a substitute for vaccination against COVID-19.
- Bamlanivimab and etesevimab are not authorized for pre-exposure prophylaxis for prevention of COVID-19.
2. Bamlanivimab and Etesevimab Injection Dosage and Administration
2.1 Patient Selection
The following medical conditions or other factors may place adults and pediatric patients, including neonates, at higher risk for progression to severe COVID-19:
- Older age (for example age ≥65 years of age)
- <1 year old
- Obesity or being overweight
- Pregnancy
- Chronic kidney disease
- Diabetes
- Immunosuppressive disease or immunosuppressive treatment
- Cardiovascular disease (including congenital heart disease) or hypertension
- Chronic lung diseases (for example, chronic obstructive pulmonary disease, asthma [moderate-to-severe], interstitial lung disease, cystic fibrosis and pulmonary hypertension)
- Sickle cell disease
- Neurodevelopmental disorders (for example, cerebral palsy) or other conditions that confer medical complexity (for example, genetic or metabolic syndromes and severe congenital anomalies)
- Having a medical-related technological dependence (for example, tracheostomy, gastrostomy, or positive pressure ventilation (not related to COVID-19))
Other medical conditions or factors (for example, race or ethnicity) may also place individual patients at high risk for progression to severe COVID-19 and authorization of bamlanivimab and etesevimab under the EUA is not limited to the medical conditions or factors listed above. For additional information on medical conditions and factors associated with increased risk for progression to severe COVID-19, see the CDC website: https://www.cdc.gov/coronavirus/2019-ncov/need-extra-precautions/people-with-medical-conditions.html. Healthcare providers should consider the benefit-risk for an individual patient.
2.2 Dosage
Treatment:
The dosage in adults (18 years and older) and pediatric patients (<18 years and weighing at least 40 kg) is bamlanivimab 700 mg and etesevimab 1,400 mg. The dosage for pediatric patients weighing less than 40 kg will vary depending on body weight:
- >20 kg to <40 kg: 350 mg bamlanivimab and 700 mg etesevimab
- >12 kg to 20 kg: 175 mg bamlanivimab and 350 mg etesevimab
- 1 kg to 12 kg: 12 mg/kg bamlanivimab and 24 mg/kg etesevimab
The recommended dosing regimen for pediatric patients ≤12 kg is predicted based on pharmacokinetic modeling and simulation [see Clinical Pharmacology (14.3)]. The youngest participant in the pediatric clinical trial for treatment was 10 months of age and weighed 8.6 kg [see Use in Specific Populations (11.3) and Clinical Trials and Supporting Data for EUA (18.1)].
For treatment of COVID-19, bamlanivimab and etesevimab should be administered together as soon as possible after positive results of direct SARS-CoV-2 viral testing and within 10 days of symptom onset.
Post-Exposure Prophylaxis:
The dosage in adults (18 years and older) and pediatric individuals (<18 years and weighing at least 40 kg) is 700 mg bamlanivimab and 1,400 mg etesevimab administered together as a single intravenous infusion. The dosage for pediatric individuals weighing less than 40 kg will vary depending on body weight:
- >20 kg to <40 kg: 350 mg bamlanivimab and 700 mg etesevimab
- >12 kg to 20 kg: 175 mg bamlanivimab and 350 mg etesevimab
- 1 kg to 12 kg: 12 mg/kg bamlanivimab and 24 mg/kg etesevimab
The recommended dosing regimen for pediatric patients ≤12 kg is predicted based on pharmacokinetic modeling and simulation [see Clinical Pharmacology (14.3)]. The youngest participant in the pediatric clinical trial for treatment was 10 months of age and weighed 8.6 kg [see Use in Specific Populations (11.3) and Clinical Trials and Supporting Data for EUA (18.1)]. Children were not enrolled in the post-exposure prophylaxis trial, BLAZE-2 [see Clinical Trials and Supporting Data for EUA (18.2)].
For post-exposure prophylaxis, bamlanivimab and etesevimab should be given together as soon as possible following exposure to SARS-CoV-2.
Under this EUA, bamlanivimab and etesevimab must be administered together as a single intravenous infusion.
2.3 Dosage Adjustment in Specific Populations
Pregnancy or Lactation
No dosage adjustment is recommended in pregnant or lactating women [see Use in Specific Populations (11.1, 11.2)].
Pediatric Use
No dosage adjustment is recommended in pediatric patients <18 years who weigh at least 40 kg. For pediatric patients weighing less than 40 kg, dosage adjustment on the basis of body weight is required [see Dosage and Administration (2.4)]. The recommended dosing regimen for pediatric patients ≤12 kg is predicted based on pharmacokinetic modeling and simulation [see Clinical Pharmacology (14.3)]. The youngest participant in the pediatric clinical trial for treatment was 10 months of age and weighed 8.6 kg [see Use in Specific Populations (11.3) and Clinical Trials and Supporting Data for EUA (18.1)]. Children were not enrolled in the post-exposure prophylaxis trial, BLAZE-2 [see Clinical Trials and Supporting Data for EUA (18.2)].
Geriatric Use
No dosage adjustment is recommended in geriatric patients [see Use in Specific Populations (11.4)].
Renal Impairment
No dosage adjustment is recommended in patients with renal impairment [see Use in Specific Populations (11.5)].
Hepatic Impairment
No dosage adjustment is recommended in patients with mild hepatic impairment. Bamlanivimab and etesevimab has not been studied in patients with moderate or severe hepatic impairment [see Use in Specific Populations (11.6)].
2.4 Dose Preparation and Administration
General Information
- Bamlanivimab and etesevimab solution for infusion should be prepared by a qualified healthcare professional using aseptic technique.
- Bamlanivimab and etesevimab are supplied in individual vials but are administered together.
- Inspect bamlanivimab and etesevimab vials visually for particulate matter and discoloration. Bamlanivimab and etesevimab are clear to opalescent and colorless to slightly yellow to slightly brown solutions.
- The prepared infusion solution should not be administered simultaneously with any other medication. The compatibility of bamlanivimab and etesevimab injection with IV solutions and medications other than 0.9% Sodium Chloride Injection is not known.
- If the infusion must be discontinued due to an infusion reaction, discard any unused product.
- The use of closed system transfer devices (CSTDs), elastomeric pumps, and pneumatic transport with bamlanivimab and etesevimab has not been studied.
- Clinically monitor patients during administration and observe patients for at least 1 hour after infusion is complete.
IV Infusion in
Adults (≥18 years regardless of weight)
and
Pediatric Patients (<18 years and weighing at least 40 kg)
Materials Needed
- 1 bamlanivimab vial (700 mg/20 mL)
- 2 etesevimab vials (700 mg/20 mL)
- 1 polyvinyl chloride (PVC) or polyethylene (PE)-line PVC, sterile prefilled infusion bag containing 0.9% Sodium Chloride Injection (sizes 50 mL to 250 mL)
- 1 PVC or PE-lined PVC infusion set
- 1 in-line or add-on 0.2/0.22 micron polyethersulfone (PES) filter
- 0.9% Sodium Chloride for flushing tubing
Preparation
- Remove 1 bamlanivimab vial and 2 etesevimab vials from refrigerated storage and allow to equilibrate to room temperature for approximately 20 minutes before preparation. Do not expose to direct heat. Do not shake the vials. Inspect vials.
- Withdraw 20 mL from one bamlanivimab vial and 40 mL from two etesevimab vials and inject all 60 mL into a prefilled infusion bag containing 0.9% Sodium Chloride (see Table 1).
- Discard any product remaining in the vials.
- Gently invert the bag by hand approximately 10 times to mix. Do not shake.
|
a 700 mg of bamlanivimab and 1,400 mg of etesevimab are added to the same infusion bag and administered together as a single intravenous infusion. |
||
|
b The minimum infusion time for patients weighing at least 40 kg and less than 50 kg who are administered bamlanivimab and etesevimab diluted in a 250-mL prefilled 0.9% Sodium Chloride infusion bag must be extended to at least 70 minutes to reduce endotoxin load. |
||
| Druga: Add 20 mL of bamlanivimab (1 vial) and 40 mL of etesevimab (2 vials) for a total of 60 mL to a prefilled infusion bag and administer as instructed below | ||
| Size of Prefilled 0.9% Sodium Chloride Infusion Bag | Maximum Infusion Rate | Minimum Infusion Time |
| 50 mL | 310 mL/hr | 21 minutes |
| 100 mL | 310 mL/hr | 31 minutes |
| 150 mL | 310 mL/hr | 41 minutes |
| 250 mL For patients weighing at least 50 kg |
310 mL/hr | 60 minutes |
| 250 mLb For patients weighing ≥40 kg and <50 kg |
266 mL/hr | 70 minutes |
Administration
- These products are preservative-free and therefore, the diluted infusion solution should be administered immediately.
- If immediate administration is not possible, store the diluted infusion solution for up to 24 hours at refrigerated temperature (2°C to 8°C [36°F to 46°F]) and up to 7 hours at room temperature (20°C to 25°C [68°F to 77°F]) including infusion time. If refrigerated, allow the infusion solution to equilibrate to room temperature for approximately 20 minutes prior to administration.
- Attach the infusion set to the IV bag. Use of in-line or add-on 0.2/0.22 micron polyethersulfone (PES) filter is strongly recommended.
- Prime the infusion set.
- Administer the entire infusion solution in the bag via pump or gravity according to the size of infusion bag used (see Table 1). Due to potential overfill of prefilled saline bags, the entire infusion solution in the bag should be administered to avoid underdosage.
- Once infusion is complete, flush the tubing with 0.9% Sodium Chloride to ensure delivery of the required dose.
IV Infusion in
Pediatric Patients (<18 years and weighing <40 kg)
Materials Needed
| IV bag | Syringe Pump |
| 1 bamlanivimab vial (700 mg/20 mL) | 1 bamlanivimab vial (700 mg/20 mL) |
| 1 etesevimab vial (700 mg/20 mL) | 1 etesevimab vial (700 mg/20 mL) |
| 1 sterile, empty 50-mL PVC or PE-lined PVC infusion bag | 1 disposable syringe |
| 1 PVC or PE-lined PVC Infusion set | 1 syringe extension set |
| 1 in-line or add-on 0.2/0.22 micron PES filter | 1 syringe pump |
| 0.9% Sodium Chloride for flushing | 0.9% Sodium Chloride for flushing |
Under this EUA, single-dose vials may be used to prepare more than one pediatric dose; in addition, pediatric doses do not need to be diluted for patients <18 years and weighing <40 kg.
Preparation
- Remove bamlanivimab and etesevimab vials from refrigerated storage and allow to equilibrate to room temperature for approximately 20 minutes before preparation. Do not expose to direct heat. Do not shake vials. Inspect vials.
- Withdraw appropriate amounts of bamlanivimab and etesevimab from vials based on body weight and inject into the empty infusion bag or draw into a disposable syringe (see Table 2).
- Multiple doses of bamlanivimab and etesevimab may be prepared from each product vial (see the storage conditions specified below). Prepare all infusion bags or syringes at the same time. Appropriately label any prepared doses including the patient weight and dose, and time of preparation to minimize risk of medication errors, particularly in cases where multiple doses are prepared simultaneously.
- Discard any product remaining in the vials after all doses have been prepared.
- Gently invert the infusion bag or syringe to mix the contents. Do not shake or vigorously agitate.
|
a Amount of BAM (as mL) and amount of ETE (as mL) for patients weighing up to 12 kg are calculated and rounded to one decimal place. |
||||
| Body Weight | BAM/ETE dose (mg) |
Amount of BAM (as mL)a |
Amount of ETE (as mL)a |
Maximum Infusion Rate |
| >20 kg to <40 kg | 350 mg / 700 mg | 10 mL | 20 mL | 1.88 mL/min |
| >12 kg to 20 kg | 175 mg / 350 mg | 5 mL | 10 mL | 0.94 mL/min |
| >11 kg to 12 kg | 138 mg / 276 mg | 3.9 mL | 7.9 mL | 0.74 mL/min |
| >10 kg to 11 kg | 126 mg / 252 mg | 3.6 mL | 7.2 mL | 0.68 mL/min |
| >9 kg to 10 kg | 114 mg / 228 mg | 3.3 mL | 6.5 mL | 0.61 mL/min |
| >8 kg to 9 kg | 102 mg / 204 mg | 2.9 mL | 5.8 mL | 0.54 mL/min |
| >7 kg to 8 kg | 90 mg / 180 mg | 2.6 mL | 5.1 mL | 0.48 mL/min |
| >6 kg to 7 kg | 78 mg / 156 mg | 2.2 mL | 4.5 mL | 0.42 mL/min |
| >5 kg to 6 kg | 66 mg / 132 mg | 1.9 mL | 3.8 mL | 0.36 mL/min |
| >4 kg to 5 kg | 54 mg / 108 mg | 1.5 mL | 3.1 mL | 0.29 mL/min |
| >3 kg to 4 kg | 42 mg / 84 mg | 1.2 mL | 2.4 mL | 0.23 mL/min |
| >2 kg to 3 kg | 30 mg / 60 mg | 0.9 mL | 1.7 mL | 0.16 mL/min |
| >1.5 kg to 2 kg | 21 mg / 42 mg | 0.6 mL | 1.2 mL | 0.11 mL/min |
| 1 kg to 1.5 kg | 15 mg / 30 mg | 0.4 mL | 0.9 mL | 0.08 mL/min |
Administration
- These products are preservative-free and therefore, the infusion solution should be administered immediately.
- If immediate administration is not possible, store the infusion solution for up to 24 hours at refrigerated temperature (2°C to 8°C [36°F to 46°F]) and up to 7 hours at room temperature (20°C to 25°C [68°F to 77°F]) including infusion time. If refrigerated, allow the infusion solution to equilibrate to room temperature for approximately 20 minutes prior to administration.
- IV bag:
- Attach the infusion set to the IV bag. Use of in-line or add-on 0.2/0.22 micron polyethersulfone (PES) filter is strongly recommended.
- Prime the infusion set.
- Administer the entire infusion solution in the bag via pump or gravity over at least 16 minutes (see Table 2).
- Once infusion is complete, flush the tubing with 0.9% Sodium Chloride to ensure delivery of the required dose.
- Syringe Pump:
- Administer the entire contents of the syringe via syringe pump over at least 16 minutes (see Table 2).
- After the entire contents of the syringe have been administered, flush the extension set with 0.9% Sodium Chloride to ensure delivery of the required dose.
3. Dosage Forms and Strengths
Bamlanivimab is a sterile, preservative-free, clear to opalescent and colorless to slightly yellow to slightly brown solution available as:
- Injection: 700 mg/20 mL (35 mg/mL) in a single-dose* vial.
Etesevimab is a sterile, preservative-free, clear to opalescent and colorless to slightly yellow to slightly brown solution available as:
- Injection: 700 mg/20 mL (35 mg/mL) in a single-dose* vial.
* Under this EUA, single-dose vials may be used to prepare more than one pediatric dose.
4. Contraindications
None.
5. Warnings and Precautions
There are limited clinical data available for bamlanivimab and etesevimab. Serious and unexpected adverse events may occur that have not been previously reported with use of bamlanivimab and etesevimab together.
5.1 Hypersensitivity Including Anaphylaxis and Infusion-Related Reactions
Serious hypersensitivity reactions, including anaphylaxis, have been observed with administration of bamlanivimab and etesevimab. If signs and symptoms of a clinically significant hypersensitivity reaction or anaphylaxis occur, immediately discontinue administration and initiate appropriate medications and/or supportive care.
Infusion-related reactions, occurring during the infusion and up to 24 hours after the infusion, have been observed with administration of bamlanivimab and etesevimab together. These reactions may be severe or life threatening.
Signs and symptoms of infusion related reactions may include [see Overall Safety Summary (6.1)]:
- fever, difficulty breathing, reduced oxygen saturation, chills, fatigue, arrhythmia (e.g., atrial fibrillation, sinus tachycardia, bradycardia), chest pain or discomfort, weakness, altered mental status, nausea, headache, bronchospasm, hypotension, hypertension, angioedema, throat irritation, rash including urticaria, pruritus, myalgia, vasovagal reactions (e.g., pre-syncope, syncope), dizziness and diaphoresis.
Consider slowing or stopping the infusion and administer appropriate medications and/or supportive care if an infusion-related reaction occurs.
Hypersensitivity reactions occurring more than 24 hours after the infusion have also been reported with the use of bamlanivimab and etesevimab under Emergency Use Authorization.
5.2 Clinical Worsening After Bamlanivimab and Etesevimab Administration
Clinical worsening of COVID-19 after administration of bamlanivimab and etesevimab together has been reported and may include signs or symptoms of fever, hypoxia or increased respiratory difficulty, arrhythmia (e.g., atrial fibrillation, sinus tachycardia, bradycardia), fatigue, and altered mental status. Some of these events required hospitalization. It is not known if these events were related to bamlanivimab and etesevimab use or were due to progression of COVID-19.
5.3 Limitations of Benefit and Potential for Risk in Patients with Severe COVID-19
Treatment with bamlanivimab and etesevimab has not been studied in patients hospitalized due to COVID-19. Monoclonal antibodies, such bamlanivimab and etesevimab, may be associated with worse clinical outcomes when administered to hospitalized patients with COVID-19 requiring high flow oxygen or mechanical ventilation. Therefore,
- Bamlanivimab and etesevimab are not authorized for use in patients 2 years and older who are hospitalized due to COVID-192,
- Bamlanivimab and etesevimab are not authorized for use in patients, regardless of age, who:
- require oxygen therapy and/or respiratory support due to COVID-19, OR
- require an increase in baseline oxygen flow rate and/or respiratory support due to COVID-19 and are on chronic oxygen therapy and/or respiratory support due to underlying non-COVID-19 related comorbidity [see Limitations of Authorized Use (1.1)].
6 OVERALL SAFETY SUMMARY
6.1 Clinical Trials Experience
Adults (≥18 Years) and Pediatric Patients (<18 Years and weighing at least 40 kg)
The safety of bamlanivimab administered with etesevimab is primarily based on exposure of approximately 1,400 ambulatory (non-hospitalized) subjects who received doses of bamlanivimab and etesevimab together, at the recommended dose or higher, in BLAZE-1 and BLAZE-4. BLAZE-1 is a Phase 2/3, randomized, double-blind, placebo-controlled clinical trial studying bamlanivimab and etesevimab administered together for the treatment of subjects with mild to moderate COVID-19. Thirty-four pediatric patients (ages 12 to <18 years and weighing at least 40 kg) were included in the Phase 3 portion of BLAZE-1 (14 received placebo, 14 received the authorized dose or a higher dose for their age, and 6 received a lower dose than authorized for their age). In the Phase 3 portion of the trial, enrolled participants had at least one risk factor for the development of severe COVID-19 illness. BLAZE-4 is a Phase 2, randomized, double-blind, placebo-controlled clinical trial studying bamlanivimab and etesevimab for the treatment of subjects with mild to moderate COVID-19. Subjects ≥65 years old or with BMI ≥35 were excluded from enrollment. In clinical trials, approximately 4,000 subjects have received bamlanivimab (either alone or with etesevimab) at doses ranging from 700 to 7,000 mg. Bamlanivimab and etesevimab at the authorized doses of 700 mg and 1,400 mg have been administered together to approximately 800 subjects in clinical trials [see Clinical Pharmacology (14.2)].
The following adverse reactions (i.e., adverse events assessed as causally related) have been observed in those who have received bamlanivimab and etesevimab together at the authorized dose or higher [see Warnings and Precautions (5.1)]:
- anaphylaxis (n=1, 0.07%)
- infusion-related reactions (n=16, 1.1%)
In the case of anaphylaxis and serious infusion-related reactions, all infusions were stopped, and treatment was administered. One case required epinephrine. All events resolved.
The most common treatment-emergent adverse events in the bamlanivimab and etesevimab treatment group in BLAZE-1 and BLAZE-4 included nausea, dizziness, and pruritus. No treatment-emergent adverse events occurred in more than 1% of participants and the rates were comparable in the treatment and placebo groups.
Pediatric Patients (Birth to <18 Years)
In addition to the 34 pediatric patients (ages 12 to <18 and weighing at least 40 kg) enrolled in the Phase 3 portion of BLAZE-1, an open-label pediatric addendum to BLAZE-1 enrolled 40 patients aged 12 to <18, 36 aged 6 to <12, 10 aged 2 to <6, and 5 birth to <2 for a total of 125 pediatric patients. All pediatric patients had at least one risk factor for the development of severe COVID-19 illness. Pediatric patients weighing 8.6 kg to <40 kg received doses of bamlanivimab and etesevimab that were adjusted for their body weight, to achieve comparable exposures as adults and adolescents receiving the authorized dosage of bamlanivimab 700 mg and etesevimab 1,400 mg, respectively. The adverse drug reaction profile in pediatric patients is consistent with the established profile.
7 PATIENT MONITORING RECOMMENDATIONS
Clinically monitor patients during administration and observe patients for at least 1 hour after infusion is complete [see Warnings and Precautions (5.1) and Overall Safety Summary (6.1)].
8. Adverse Reactions/Side Effects
Clinical trials evaluating the safety of bamlanivimab and etesevimab are ongoing [see Overall Safety Summary (6)].
Completion of FDA MedWatch Form to report all medication errors and serious adverse events* occurring during bamlanivimab and etesevimab use and considered to be potentially related to bamlanivimab and etesevimab is mandatory and must be done by the prescribing healthcare provider and/or the provider’s designee. These adverse events must be reported within 7 calendar days from the onset of the event:
*Serious adverse events are defined as:
- death;
- a life-threatening adverse event;
- inpatient hospitalization or prolongation of existing hospitalization;
- a persistent or significant incapacity or substantial disruption of the ability to conduct normal life functions;
- a congenital anomaly/birth defect;
- a medical or surgical intervention to prevent death, a life-threatening event, hospitalization, disability, or congenital anomaly.
If a serious and unexpected adverse event occurs and appears to be associated with the use of bamlanivimab and etesevimab under this EUA, the prescribing healthcare provider and/or the provider’s designee must complete and submit a MedWatch form to FDA using one of the following methods:
- Complete and submit the report online: www.fda.gov/medwatch/report.htm, or
- Complete and submit a postage-paid FDA Form 3500 (https://www.fda.gov/media/76299/download) and return by:
- Mail to MedWatch, 5600 Fishers Lane, Rockville, MD 20852-9787, or
- Fax (1-800-FDA- 0178), or
- Call 1-800-FDA-1088 to request a reporting form
IMPORTANT: When reporting adverse events or medication errors to MedWatch, please complete the entire form with detailed information. It is important that the information reported to FDA be as detailed and complete as possible. Information that must be included:
- Patient demographics (e.g., patient initials, date of birth)
- Pertinent medical history
- Pertinent details regarding adverse events and course of illness
- Concomitant medications
- Timing of adverse event(s) in relationship to administration of bamlanivimab and etesevimab
- Pertinent laboratory and virology information
- Outcome of the event and any additional follow-up information if it is available at the time of the MedWatch report. Subsequent reporting of follow-up information should be completed if additional details become available.
The following steps are highlighted to provide the necessary information for safety tracking:
- In section A, box 1, provide the patient’s initials in the Patient Identifier
- In section A, box 2, provide the patient’s date of birth
- In section B, box 5, description of the event:
- Write “bamlanivimab and etesevimab use for COVID-19 under Emergency Use Authorization (EUA)” as the first line
- Provide a detailed report of medication error and/or adverse event. It is important to provide detailed information regarding the patient and adverse event/medication error for ongoing safety evaluation of this unapproved drug. Please see information to include listed above.
- In section G, box 1, name and address:
- Provide the name and contact information of the prescribing healthcare provider or institutional designee who is responsible for the report.
- Provide the address of the treating institution (NOT the healthcare provider’s office address).
9 OTHER REPORTING REQUIREMENTS
- Healthcare facilities and providers must report therapeutics information and utilization data through HHS Protect, Teletracking or National Healthcare Safety Network (NHSN) as directed by the U.S. Department of Health and Human Services.
- In addition, please provide a copy of all FDA MedWatch forms to:
Eli Lilly and Company, Global Patient Safety
Fax: 1-317-277-0853
E-mail: mailindata_gsmtindy@lilly.com
Or call Eli Lilly and Company at 1-855-LillyC19 (1-855-545-5921) to report adverse events.
10. Drug Interactions
Bamlanivimab and etesevimab are not renally excreted or metabolized by cytochrome P450 enzymes; therefore, interactions with concomitant medications that are renally excreted or that are substrates, inducers, or inhibitors of cytochrome P450 enzymes are unlikely.
11. Use In Specific Populations
11.1 Pregnancy
Risk Summary
There are insufficient data to evaluate a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. Bamlanivimab and etesevimab should only be used during pregnancy if the potential benefit outweighs the potential risk for the mother and the fetus. There are maternal and fetal risks associated with untreated COVID-19 in pregnancy (see Clinical Considerations).
Nonclinical reproductive toxicity studies have not been performed with bamlanivimab or etesevimab. In tissue cross reactivity studies using human fetal tissues, no binding of clinical concern was detected for etesevimab or bamlanivimab. Human immunoglobulin G1 (IgG1) antibodies are known to cross the placental barrier; therefore, bamlanivimab and etesevimab have the potential to be transferred from the mother to the developing fetus. It is unknown whether the potential transfer of bamlanivimab or etesevimab provides any treatment benefit or risk to the developing fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo-Fetal Risk
COVID-19 in pregnancy is associated with adverse maternal and fetal outcomes, including preeclampsia, eclampsia, preterm birth, premature rupture of membranes, venous thromboembolic disease, and fetal death.
11.2 Lactation
Risk Summary
There are no available data on the presence of bamlanivimab or etesevimab in human or animal milk, the effects on the breastfed infant, or the effects on milk production. Maternal IgG is known to be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for bamlanivimab and etesevimab and any potential adverse effects on the breastfed child from bamlanivimab and etesevimab or from the underlying maternal condition. Breastfeeding individuals with COVID-19 should follow practices according to clinical guidelines to avoid exposing the infant to COVID-19.
11.3 Pediatric Use
Bamlanivimab and etesevimab administered together are authorized for the treatment of mild to moderate COVID-19 and post-exposure prophylaxis for prevention of COVID-19 in pediatric patients, including neonates [see Authorized Use (1)]. Given the similar course of COVID-19, the authorization of bamlanivimab and etesevimab for treatment and post-exposure prophylaxis in younger pediatric patients, including neonates, is supported by safety and efficacy data in adolescents and adults, together with additional pharmacokinetic and safety data from the clinical trial in pediatric patients studying bamlanivimab and etesevimab for the treatment of mild to moderate COVID-19.
Use of bamlanivimab and etesevimab in pediatric patients is based on analyses of data from BLAZE-1 in subjects aged 10 months to 18 years of age [see Clinical Pharmacology (14.3) and Clinical Trials and Supporting Data for EUA (18.1)]. No dosage adjustment is recommended in pediatric patients 12-18 years of age who weigh at least 40 kg. Pediatric patients weighing less than 40 kg should be dosed on the basis of body weight [see Dosage and Administration (2.2, 2.4)]. The recommended dosing regimen for pediatric patients ≤12 kg is predicted based on pharmacokinetic modeling and simulation [see Clinical Pharmacology (14.3)]. The youngest participant in the pediatric clinical trial for treatment was 10 months of age and weighed 8.6 kg [see Clinical Trials and Supporting Data for EUA (18.1)]. Safety in pediatric patients was similar to what was observed in adults [see Clinical Trial Experience (6.1)]. Children were not enrolled in the post-exposure prophylaxis trial, BLAZE-2 [see Clinical Trials and Supporting Data for EUA (18.2)].
11.4 Geriatric Use
Of the 1141 patients receiving bamlanivimab and etesevimab in BLAZE-1, 30% were 65 years of age and older and 10% were 75 years of age and older. Based on population PK analyses, there is no difference in PK of bamlanivimab or etesevimab in geriatric patients compared to younger patients [see Clinical Trial Results and Supporting Data for EUA (18.1)].
11.5 Renal Impairment
Bamlanivimab and etesevimab are not eliminated intact in the urine, thus renal impairment is not expected to affect the exposure of bamlanivimab or etesevimab.
11.6 Hepatic Impairment
Based on population PK analysis, there is no difference in PK of bamlanivimab or etesevimab in patients with mild hepatic impairment compared to patients with normal hepatic function. Bamlanivimab and etesevimab have not been studied in patients with moderate or severe hepatic impairment.
11.7 Other Specific Populations
Based on population PK analysis, the PK of bamlanivimab and etesevimab was not affected by sex, race, or disease severity. Body weight had no clinically relevant effect on the PK of bamlanivimab and etesevimab in adults with COVID-19 over the body weight range of 41 kg to 173 kg.
12. Overdosage
Doses up to 7,000 mg of bamlanivimab (10 times the authorized dose of bamlanivimab for adults [≥18 years] and pediatric patients [<18 years weighing at least 40 kg]) or 7,000 mg of etesevimab (5 times the authorized dose of etesevimab for adults [≥18 years] and pediatric patients [<18 years weighing at least 40 kg]) have been administered in clinical trials without dose-limiting toxicity. Treatment of overdose with bamlanivimab and etesevimab should consist of general supportive measures including monitoring of vital signs and observation of the clinical status of the patient. There is no specific antidote for overdose with either bamlanivimab or etesevimab.
13. Bamlanivimab and Etesevimab Injection Description
Bamlanivimab
Bamlanivimab is a human immunoglobulin G-1 (IgG1 variant) monoclonal antibody consisting of 2 identical light chain polypeptides composed of 214 amino acids each and 2 identical heavy chain polypeptides composed of 455 amino acids produced by a Chinese Hamster Ovary (CHO) cell line and molecular weight of 146 kDa.
Bamlanivimab injection is a sterile, preservative-free, clear to opalescent and colorless to slightly yellow to slightly brown solution in a vial for intravenous infusion.
Each mL contains 35 mg of bamlanivimab, and L-histidine (0.4 mg), L-histidine hydrochloride monohydrate (0.6 mg), sodium chloride (2.9 mg), sucrose (60 mg), polysorbate 80 (0.5 mg), and Water for Injection. The bamlanivimab solution has a pH range of 5.5-6.5.
Etesevimab
Etesevimab is a human IgG1 variant monoclonal antibody (mAb) consisting of 2 identical light chain polypeptides composed of 216 amino acids each and 2 identical heavy chain polypeptides composed of 449 amino acids produced by a Chinese Hamster Ovary (CHO) cell line and molecular weight of 145 kDa.
Etesevimab injection is a sterile, preservative-free, clear to opalescent and colorless to slightly yellow to slightly brown solution in a vial for intravenous infusion.
Each mL contains 35 mg of etesevimab, L-histidine (1.55 mg), L-histidine hydrochloride monohydrate (2.10 mg), sucrose (80.4 mg), polysorbate 80 (0.5 mg), and Water for injection. The etesevimab solution has a pH range of 5.5.-6.5.
14. Bamlanivimab and Etesevimab Injection — Clinical Pharmacology
14.1 Mechanism of Action
Bamlanivimab is a recombinant neutralizing human IgG1ϰ monoclonal antibody (mAb) to the spike protein of SARS-CoV-2 and is unmodified in the Fc region. Bamlanivimab binds the spike protein with a dissociation constant KD = 0.071 nM and blocks spike protein attachment to the human ACE2 receptor with an IC50 value of 0.17 nM (0.025 μg/mL).
Etesevimab is a recombinant neutralizing human IgG1ϰ mAb to the spike protein of SARS-CoV-2, with amino acid substitutions in the Fc region (L234A, L235A) to reduce effector function. Etesevimab binds the spike protein with a dissociation constant KD = 6.45 nM and blocks spike protein attachment to the human ACE2 receptor with an IC50 value of 0.32 nM (0.046 μg/mL).
Bamlanivimab and etesevimab bind to different but overlapping epitopes in the receptor binding domain (RBD) of the S-protein. Using both antibodies together is expected to reduce the risk of viral resistance.
14.2 Pharmacodynamics
A flat exposure-response relationship for efficacy was identified for bamlanivimab and etesevimab administered together within the dose range of 700 mg bamlanivimab and 1,400 mg etesevimab to 2,800 mg bamlanivimab and 2,800 mg etesevimab (4 and 2 times the authorized dose, respectively), based on clinical data and pharmacokinetic/pharmacodynamic modeling.
For post-exposure prophylaxis of COVID-19, a dose of 700 mg bamlanivimab and 1,400 mg etesevimab was supported based on clinical data and pharmacokinetic/pharmacodynamic modeling.
14.3 Pharmacokinetics
A summary of PK parameters of bamlanivimab and etesevimab following administration of a single dose of 700 mg bamlanivimab and 1,400 mg etesevimab is provided in Table 3. There is no change in PK of bamlanivimab or etesevimab administered alone or together suggesting there is no interaction between the two antibodies. There were no differences in PK of etesevimab between mild/moderate ambulatory participants and healthy participants.
|
Abbreviations: CV = coefficient of variation; Cmax = maximum concentration; AUCinf = area under the concentration versus time curve from zero to infinity; Vss = steady-state volume of distribution. |
|||
|
a N = number of subjects simulated using the PK model. |
|||
|
b The number of subjects for Vss, half-life, and clearance are based on a population PK model that included bamlanivimab doses up to 7,000 mg and etesevimab doses up to 2,800 mg. |
|||
| N | BAM (700 mg) |
ETE (1400 mg) |
|
| Systemic Exposure | |||
| Geometric Mean (%CV) Cmax, mcg/mL | 270 | 187 (41.7) | 422 (41.2) |
| Geometric Mean (%CV) Cday 29, mcg/mL | 311 BAM; 320 ETE | 25.7 (42.9) | 116 (38.1) |
| Median (5th,95th percentile) Cweek 8, mcg/mL | 1000a | 10.1 (3.59, 22.9) | 58.3 (26.8, 117) |
| Geometric Mean (%CV) AUCinf, mcg day/mL | 499 | 2500 (28.0) | 10600 (29.9) |
| Distribution | |||
| Geometric Mean (%CV) Vss (L) | 1899 BAM; 1498 ETEb | 6.59 (24.9) | 5.78 (24.7) |
| Elimination | |||
| Geometric Mean (%CV) Elimination Half-Life (day) | 1899 BAM; 1498 ETEb | 20.9 (17.3) | 32.6 (21.7) |
| Geometric Mean (%CV) Clearance (L/day) | 1899 BAM; 1498 ETEb | 0.274 (31.5) | 0.134 (32.5) |
Bamlanivimab and etesevimab are expected to be degraded into small peptides and component amino acids via catabolic pathways in the same manner as endogenous IgG antibodies.
Special Populations:
The PK profiles of bamlanivimab and etesevimab were not affected by age, sex, race, or disease severity based on a population PK analysis. Body weight had no clinically relevant effect on the PK of bamlanivimab or etesevimab in adults with COVID-19 over the body weight range of 41 kg to 173 kg [see Use in Specific Populations (11.4, 11.7)].
Pediatric population
The PK of bamlanivimab and etesevimab has been evaluated in 88 pediatric patients <18 years who received weight-based dosing [see Dosage and Administration (2.2)]. The data show that weight-based dosing in pediatric patients provides comparable plasma exposures to those observed in adults who received bamlanivimab 700 mg and etesevimab 1,400 mg. No dosage adjustment is recommended in pediatric patients <18 years who weigh at least 40 kg. Pediatric patients weighing less than 40 kg should be dosed on the basis of body weight [see Dosage and Administration (2.2, 2.4)]. The recommended dosing regimen for pediatric patients ≤12 kg is predicted to result in similar exposures when compared to exposures achieved in adults receiving bamlanivimab 700 mg and etesevimab 1,400 mg based on pharmacokinetic modeling and simulation. The youngest participant in the pediatric treatment trial was 10 months of age and weighed 8.6 kg [see Clinical Trials and Supporting Data for EUA (18.1)].
|
Abbreviations: CV = coefficient of variation; Cmax = maximum concentration; AUCinf = area under the concentration versus time curve from zero to infinity. |
||||
| Body Weight | ≥40 kg | >20 to <40 kg | >12 to ≤20 kg | ≤12 kg |
| BAM / ETE Dose | 700 mg / 1400 mg | 350 mg / 700 mg | 175 mg / 350 mg | 15 mg/kg / 30 mg/kg |
| BAM: Geometric Mean (%CV) [n] | ||||
| Cmax, mcg/mL | 235 (51.0) [52] | 239 (39.1) [16] | 243 (66.0) [7] | 371 (9.8) [2] |
| Cday 29, mcg/mL | 26.8 (50.2) [34] | 26.1 (32.5) [8] | 23.0 (53.0) [3] | [0] |
| AUCinf, mcg day/mL | 2760 (30.7) [66] | 2780 (25.7) [20] | 2430 (28.4) [9] | 3000 (19.1) [3] |
| ETE: Geometric Mean (%CV) [n] | ||||
| Cmax, mcg/mL | 508 (50.6) [50] | 444 (26.6) [14] | 444 (64.9) [7] | 831 (16.8) [2] |
| Cday 29, mcg/mL | 133 (46.8) [34] | 138 (29.5) [8] | 125 (51.5) [3] | [0] |
| AUCinf, mcg day/mL | 12900 (32.4) [66] | 12400 (23.2) [20] | 11300 (29.6) [9] | 13500 (13.0) [3] |
Patients with renal impairment
Bamlanivimab and etesevimab are not eliminated intact in the urine. Renal impairment is not expected to impact the PK of bamlanivimab and etesevimab, since mAbs with molecular weight >69 kDa are known not to undergo renal elimination. Similarly, dialysis is not expected to impact the PK of bamlanivimab and etesevimab [see Use in Specific Populations (11.5)].
Patients with hepatic impairment
Based on population PK analysis, there is no significant difference in PK of bamlanivimab or etesevimab in patients with mild hepatic impairment compared to patients with normal hepatic function. Bamlanivimab and etesevimab have not been studied in patients with moderate or severe hepatic impairment [see Use in Specific Populations (11.6)].
Drug interactions:
Bamlanivimab and etesevimab are not renally excreted or metabolized by cytochrome P450 enzymes; therefore, interactions with concomitant medications that are renally excreted or that are substrates, inducers, or inhibitors of cytochrome P450 enzymes are unlikely.
15 MICROBIOLOGY/RESISTANCE INFORMATION
Antiviral Activity
The cell culture neutralization activity of bamlanivimab and of etesevimab against SARS-CoV-2 was measured in a dose-response model quantifying plaque reduction using cultured Vero E6 cells. Bamlanivimab, etesevimab and a 1:1 (weight/weight) ratio of bamlanivimab and etesevimab together neutralized the USA/WA/1/2020 isolate of SARS-CoV-2 with estimated EC50 values = 0.14 nM (0.02 μg/mL), 0.97 nM (0.14 μg/mL) and 0.14 nM (0.02 μg/mL), respectively.
Bamlanivimab demonstrated antibody-dependent cell-mediated cytotoxicity on reporter Jurkat cells expressing FcγRIIIa following engagement with target cells expressing spike protein. Bamlanivimab did not elicit complement-dependent cytotoxicity activity in cell-based assays.
Etesevimab did not demonstrate detectable antibody-dependent cell-mediated cytotoxicity on Jurkat reporter cells expressing FcγRIIIa. Etesevimab did not elicit complement-dependent cytotoxicity activity in cell-based assays.
Antibody Dependent Enhancement (ADE) of Infection
The risk that bamlanivimab and etesevimab could mediate viral uptake and replication by immune cells was studied in THP-1 and Raji cell lines and primary human macrophages. In general, experiments with bamlanivimab, with etesevimab, and with bamlanivimab and etesevimab together did not demonstrate productive viral infection in immune cells exposed to SARS-CoV-2 at concentrations of mAb(s) down to at least 100-fold below the respective EC50 value(s).
Antiviral Resistance
There is a potential risk of treatment failure due to the development of viral variants that are resistant to bamlanivimab and/or etesevimab (Table 5). There are other authorized treatments available and healthcare providers should choose an authorized therapeutic option with activity against circulating variants in their state, territory, or US jurisdiction. Variant frequency data for states, territories, and US jurisdictions can be accessed on the following CDC website: https://www.cdc.gov/coronavirus/2019-ncov/cases-updates/variant-proportions.html.
Resistant variants were identified using directed evolution of the spike protein and serial passage in cell culture of SARS-CoV-2 in the presence of bamlanivimab or etesevimab individually. Resistant variants were not identified when bamlanivimab and etesevimab were tested together using the same methodology. Viral variants identified in these studies that had reduced susceptibility to bamlanivimab included spike protein amino acid substitutions E484D/K/Q, F490S, Q493R, and S494P, and variants that had reduced susceptibility to etesevimab included substitutions K417N, D420N, and N460K/S/T/Y. Neutralization assays using SARS-CoV-2 and vesicular stomatitis virus (VSV) virus-like particles (VLP) pseudotyped with variant SARS-CoV-2 spike protein confirmed reductions in susceptibility to the selecting antibody. Retention of susceptibility to the other antibody alone was observed, with the exception of the E484D and Q493R substitution. All variants maintained susceptibility to bamlanivimab and etesevimab together, with the exception of those with E484D, E484K, E484Q, and Q493R substitutions, which had reduced susceptibility of 145-fold, 24-fold, 17-fold, and 1,054-fold, respectively in a pseudotyped VLP assay.
Evaluation of susceptibility of variants identified through global surveillance in subjects treated with bamlanivimab and etesevimab is ongoing. Pseudotyped VLP evaluation of amino acid substitutions identified in global surveillance showed that the V483A substitution reduced susceptibility to bamlanivimab 48-fold, but activity was maintained with etesevimab, and with bamlanivimab and etesevimab together. N501Y and N501T substitutions reduced susceptibility to etesevimab approximately 5-fold and 20-fold, respectively. Activity against variants with N501Y or N501T substitutions was maintained with bamlanivimab alone, and with bamlanivimab and etesevimab together.
Bamlanivimab and etesevimab together retained activity against a SARS-CoV-2 B.1.1.7 lineage (Alpha; UK origin) virus and related pseudotyped VLPs expressing the spike protein found in the B.1.1.7 variant (Tables 5 and 6). SARS-CoV-2 B.1.351 lineage (Beta; South Africa origin) virus and related pseudotyped VLPs expressing spike proteins from B.1.351 lineage or substitutions K417N + E484K + N501Y found in this lineage had reduced susceptibility to bamlanivimab and etesevimab together of >324, 431-fold or >45-fold, respectively. Pseudotyped VLPs expressing spike protein from the P.1 lineage (Gamma; Brazil origin) or K417T + E484K + N501Y found in the P.1 lineage had reduced susceptibility to bamlanivimab and etesevimab together of 252-fold or >3,351-fold, respectively.
Bamlanivimab and etesevimab together and etesevimab alone retained activity against SARS-CoV-2 B.1.617.2 lineage (Delta; India origin) virus and related pseudotyped VLPs, but bamlanivimab alone had reduced activity (>1,136 and >1,868-fold, respectively). Bamlanivimab and etesevimab are expected to retain activity against B.1.617.2 sublineage AY.3 (India origin). B.1.617.2 sublineages AY.1/AY.2 (India origin) have an additional K417N substitution; pseudotyped VLPs expressing AY.1/AY.2 related spike sequence had a reduced susceptibility to bamlanivimab and etesevimab together of 1,235-fold. SARS-CoV-2 recombinant virus containing the L452R substitution present in B.1.427/B.1.429 lineages (Epsilon; USA [California] origin) and pseudotyped VLPs expressing the full-length spike protein or the L452R substitution found in this lineage showed reduced susceptibility to bamlanivimab and etesevimab together of 11-fold, 9-fold or 5-fold, respectively. Pseudotyped VLPs expressing spike protein from the B.1.617.1 lineage (Kappa; India origin) showed reduced susceptibility to bamlanivimab and etesevimab together of 6-fold; for this variant, susceptibility to etesevimab alone was maintained, but not to bamlanivimab alone (>1,030-fold reduction). Bamlanivimab and etesevimab together and etesevimab alone retained activity against pseudotyped VLPs expressing the full-length spike protein from the C.37 lineage (Lambda; Peru origin), but bamlanivimab alone had reduced activity (>2,112-fold reduction). Pseudotyped VLPs expressing spike protein from the B.1.621 lineage (Mu; Colombia origin) show reduced susceptibility to bamlanivimab and etesevimab together of 116-fold, due to susceptibility reductions to bamlanivimab (>1,863-fold) and etesevimab (17-fold) alone. Pseudotyped VLPs expressing the spike protein from the B.1.1.529/BA.1 lineage (Omicron; South Africa origin) show reduced susceptibility to bamlanivimab alone (>1,465-fold), etesevimab alone (>616-fold), and bamlanivimab and etesevimab together (>2,938-fold).
|
a Key substitutions occurring in the receptor binding domain of spike protein are listed. Pseudoviruses containing the full-length spike protein reflective of the consensus sequence for each of the variant lineages were tested. |
||||
|
b No change: <5-fold reduction in susceptibility. |
||||
|
c Bamlanivimab and etesevimab together are unlikely to be active against variants from this lineage. |
||||
|
d Etesevimab retains activity against this variant. |
||||
|
e Isolates of the B.1.526 lineage harbor several spike protein amino acid substitutions, and not all isolates contain the E484K substitution (as of February 2021). |
||||
| Lineage with Spike Protein Substitution | Country First Identified | WHO Nomenclature | Key Substitutions Testeda | Fold Reduction in Susceptibility |
| B.1.1.7 | UK | Alpha | N501Y | no changeb |
| B.1.351 | South Africa | Beta | K417N + E484K + N501Y | 431c |
| P.1 | Brazil | Gamma | K417T + E484K + N501Y | 252c |
| B.1.617.2/AY.3 | India | Delta | L452R + T478K | no changeb |
| AY.1/AY.2 (B.1.617.2 sublineages) |
India | Delta [+K417N] | L452R + T478K + K417N | 1,235c |
| B.1.427/B.1.429 | USA (California) | Epsilon | L452R | 9d |
| B.1.526e | USA (New York) | Iota | E484K | 30 |
| B.1.617.1 | India | Kappa | L452R + E484Q | 6d |
| C.37 | Peru | Lambda | L452Q + F490S | no changeb |
| B.1.621 | Colombia | Mu | R346K + E484K + N501Y | 116c |
| B.1.1.529/BA.1 | South Africa | Omicron | G339D + S371L + S373P + S375F + K417N + N440K + G446S + S477N + T478K + E484A + Q493R + G493S + Q498R + N501Y + Y505H | >2,938c |
|
a The B.1.1.7 variant was assessed using cell culture-expanded virus isolates and tested using an immunofluorescence based microneutralization assay and by plaque reduction assay; B.1.351 and B.1.617.2 variants were assessed using cell culture-expanded virus isolates and tested using a plaque reduction assay; the B.1.526/E484K and B.1.427/B.1.429/L452R substitutions were assessed using recombinant SARS-CoV-2 (USA/WA/1/2020 isolate with E484K or L452R) and tested using a plaque reduction assay. |
||||
|
b Key substitutions occurring in receptor binding domain of spike protein which are associated with each lineage. |
||||
|
c No change: <5-fold reduction in susceptibility. |
||||
|
d Isolates of the B.1.526 lineage harbor several spike protein amino acid substitutions, and not all isolates contain the E484K substitution (as of February 2021). This assay was conducted using recombinant SARS-CoV-2 with the E484K substitution only. |
||||
| Lineage with Spike Protein Substitution | Country First Identified | WHO Nomenclature | Key Substitutions Testedb | Fold Reduction in Susceptibility |
| B.1.1.7 | UK | Alpha | N501Y | no changec |
| B.1.351 | South Africa | Beta | K417N, E484K, N501Y | >324 |
| B.1.617.2/AY.3 | India | Delta | L452R, T478K | no changec |
| B.1.427/B.1.429 | USA (California) | Epsilon | L452R | 11 |
| B.1.526d | USA (New York) | Iota | E484K | 11 |
Due to the large reduction of pseudotyped VLP neutralization activity of both bamlanivimab and etesevimab against the substitutions in B.1.351 (Beta; South Africa origin), P.1 (Gamma; Brazil origin), AY.1/AY.2 (Delta [+K417N]; India origin), B.1.621 (Mu; Colombia origin), and B.1.1.529/BA.1 (Omicron; South Africa origin), it is unlikely that bamlanivimab and etesevimab together will be active against these variants.
It is unclear how small reductions in susceptibility to bamlanivimab and etesevimab seen in authentic or recombinant SARS-CoV-2 or pseudotyped VLP assays correlate with clinical outcomes.
In authentic SARS-CoV-2 assays, bamlanivimab and etesevimab together retained activity against variants of B.1.1.7 (Alpha) and B.1.617.2/AY.3 (Delta) lineages (Table 6), although bamlanivimab alone had reduced activity to B.1.617.2/AY.3 (Delta) in this assay (>1,136-fold). SARS-CoV-2 (USA/WA/1/2020 isolate) engineered to express the E484K substitution present in the B.1.526 lineage (Iota; USA [New York] origin) or the L452R substitution present in the B.1.427/B.1.429 lineage (Epsilon; USA [California] origin) showed reduced susceptibility to bamlanivimab and etesevimab together of 11-fold. Susceptibility to etesevimab alone was maintained for both isolates, but not to bamlanivimab alone (>833-fold and >1,460-fold reduction for E484K and L452R viruses, respectively). Available nonclinical and clinical PK data indicate that etesevimab at the authorized dose may retain activity against the B.1.526 variant clinically, although only very limited data are currently available from patients infected with this variant in clinical trials. Preliminary clinical evidence indicates that the administration of bamlanivimab and etesevimab together result in similar viral load reductions in participants infected with the L452R variant (Epsilon; USA [California] origin) as observed in those who were infected with bamlanivimab-sensitive strains. Of the 134 participants infected with the L452R variant at baseline in the Phase 3 portion of BLAZE-1, 3 of the 50 individuals treated with placebo (6%) and 1 of the 84 participants treated with bamlanivimab 700 mg and etesevimab 1,400 mg (1%) were hospitalized (p=0.15).
Genotypic and phenotypic testing are ongoing to monitor for potential bamlanivimab- and etesevimab-resistance associated spike variations in clinical trials. Analysis of baseline samples show that 8.4% (188/2246) of clinical trial patients were infected with viral variants containing single amino acid substitutions at positions associated with reduced susceptibility to either bamlanivimab or etesevimab as predicted by pseudotyped VLP or authentic SARS-CoV-2 neutralization assays. No patients were infected with a variant that was predicted to have reduced susceptibility to both bamlanivimab and etesevimab by these assessments.
Patient samples were also analyzed for treatment-emergent viral variants, defined as variants with single amino acid substitutions at positions that had reduced susceptibility to either bamlanivimab or etesevimab present at an allele fraction of ≥15%.
- In the Phase 3 portion of BLAZE-1, treatment-emergent variants were observed in 9.0% (42/467) of patients treated with bamlanivimab 2,800 mg and etesevimab 2,800 mg together, in 5.3% (21/394) of patients treated with bamlanivimab 700 mg and etesevimab 1,400 mg together, and in 4.0% (27/674) of patients treated with placebo. The majority of these were only detected at one time point in the sequential series with 0.9% (4/467), 1.0% (4/394), and 0.3% (2/674) of patients having multiple instances of detection in the bamlanivimab 2,800 mg and etesevimab 2,800 mg together, bamlanivimab 700 mg and etesevimab 1,400 mg together, and placebo groups, respectively.
- In patients treated with bamlanivimab and etesevimab together, substitutions detected in one or more patients included ones with reduced susceptibility (≥5-fold) to bamlanivimab only: L452R/W, E484K, G485V, F490L, and S494P; and ones with reduced susceptibility to etesevimab only: D405G/Y, K417N, D420N/Y, N460H/I/T, A475S/V, Y489H, and N501I/Y. While these variants had reduced susceptibility to either bamlanivimab OR etesevimab compared to wild-type in a pseudotyped VSV VLP or authentic virus assay they still retained susceptibility to the other antibody in the combination.
- There were also observations of variants with reduced susceptibility (≥5-fold) to both bamlanivimab and etesevimab and to bamlanivimab + etesevimab tested together: E484D (n=1; 145-fold reduction to bamlanivimab + etesevimab tested together at a molar ratio of 1:2), Q493K/R (n=9; 584-fold and 1,054-fold reduction to bamlanivimab + etesevimab tested together at a molar ratio of 1:2 for Q493K and Q493R, respectively) out of a total of 861 patients treated with bamlanivimab and etesevimab together.
- In a subgroup of participants infected with virus harboring L452R substitution found in the B.1.427/B.1.429 (Epsilon) lineage, a S459P treatment-emergent substitution was identified in one subject. Concurrent L452R+S459P substitutions conferred a 1,656-fold reduction in susceptibility to bamlanivimab + etesevimab together (1:2 molar ratio).
- Additional treatment-emergent substitutions in patients treated with bamlanivimab and etesevimab together, with no phenotypic data, include D405del, D420G, C480R, G485D, S494L, and P499L. The impact of these substitutions on susceptibility is not currently known.
- In a subgroup of 53 pediatric subjects who were infected with a B.1.617.2 (Delta)-related variant, which has reduced susceptibility to bamlanivimab (>1,136-fold), the following treatment-emergent substitutions with reduced susceptibility to etesevimab were detected: D420A (n=2), N460T (n=1), N460Y (n=1). Three of these four subjects had high viral load (>5.27 log10) on Day 7.
- Additional treatment-emergent substitutions with no phenotypic data detected in other pediatric subjects who were infected with a B.1.617.2 (Delta)-related variant at an allele fraction of ≥50% included: F347C, V401L, G431S and I434V.
It is possible that bamlanivimab and etesevimab resistance-associated variants could have cross-resistance to other mAbs targeting the receptor binding domain of SARS-CoV-2. The clinical impact is not known.
Immune Response Attenuation
There is a theoretical risk that antibody administration may attenuate the endogenous immune response to SARS-CoV-2 and make patients more susceptible to re-infection.
16. Nonclinical Toxicology
Carcinogenesis, mutagenesis, and reproductive toxicology studies with bamlanivimab or etesevimab have not been conducted.
In toxicology studies, bamlanivimab and etesevimab had no adverse effects when administered intravenously to rats and monkeys, respectively. Non-adverse increases in neutrophils were observed in rats dosed with bamlanivimab.
In tissue cross reactivity studies using human adult and fetal tissues, no binding of clinical concern was detected for bamlanivimab or etesevimab.
17 ANIMAL PHARMACOLOGIC AND EFFICACY DATA
Antiviral Activity In Vivo
Prophylactic administration of bamlanivimab to female Rhesus macaques (n=3 or 4 per group) resulted in 1 to 4 log10 decreases in viral genomic RNA and viral replication (sub-genomic RNA) in bronchoalveolar lavage samples relative to control animals, but less of an impact on viral RNA in throat and nasal swabs following SARS-CoV-2 inoculation.
Prophylactic or therapeutic administration of etesevimab to male Rhesus macaques (n=3 per group) resulted in approximately 4 or 3 log10 average decreases, respectively, in viral genomic RNA in oropharyngeal swabs at Day 4 post infection relative to control animals.
The applicability of these findings to a prophylaxis or treatment setting is not known.
18 CLINICAL TRIAL RESULTS AND SUPPORTING DATA FOR EUA
18.1 Treatment of Mild to Moderate COVID-19 (BLAZE-1)
Adults (≥18 Years) and Pediatric Patients (12 to <18 Years Weighing at Least 40 kg)
The data supporting this EUA for treatment of mild to moderate COVID-19 are primarily based on analyses of data from the Phase 2/3 BLAZE-1 trial (NCT04427501). This trial provides Phase 3 placebo-controlled clinical efficacy data from subjects receiving 700 mg bamlanivimab and 1,400 mg of etesevimab together, as well as for subjects receiving 2,800 mg bamlanivimab and 2,800 mg etesevimab together.
BLAZE-1 is a randomized, double-blind, placebo-controlled clinical trial studying bamlanivimab and etesevimab administered together for the treatment of subjects with mild to moderate COVID-19 (subjects with COVID-19 symptoms who are not hospitalized). BLAZE-1 enrolled adult subjects who were not hospitalized and had at least 1 or more COVID-19 symptoms that were at least mild in severity. Treatment was initiated within 3 days of obtaining the clinical sample for the first positive SARS-CoV-2 viral infection determination. Subjects in the Phase 3 portion of the trial met the criteria for high-risk (as defined in Section 2).
Phase 3 Data from BLAZE-1 (bamlanivimab 700 mg and etesevimab 1,400 mg)
In this portion of the trial, subjects were treated with a single infusion of bamlanivimab 700 mg and etesevimab 1,400 mg (N=511) or placebo (N=258). The majority (99.2%) of the patients enrolled in these dose arms met the criteria for high-risk adults (≥18 years of age) that included at least one of the following: age ≥65 years, BMI ≥35, chronic kidney disease, diabetes, immunosuppressive disease, immunosuppressant treatment, or age ≥55 years with cardiovascular disease, hypertension, chronic pulmonary disease or other chronic respiratory disease. Participants ages 12-17 were also enrolled in the trial (10 [2.0%] were treated with bamlanivimab and etesevimab and 13 [1.7%] were treated with placebo), and met high-risk criteria as defined in the trial protocol.
At baseline, median age was 56 years (with 30% of subjects aged 65 or older); 53% of subjects were female, 87% were White, 27% were Hispanic or Latino, and 8% were Black or African American. Subjects had mild (76%) to moderate (24%) COVID-19; the mean duration of symptoms was 4 days; mean viral load by cycle threshold (CT) was 24.33 at baseline. The baseline demographics and disease characteristics were well balanced across treatment groups.
The primary endpoint was the proportion of subjects with COVID-19 related hospitalization (defined as ≥24 hours of acute care) or death by any cause by Day 29. Events occurred in 15 subjects treated with placebo (6%) as compared to 4 events in subjects treated with bamlanivimab 700 mg and etesevimab 1,400 mg together (0.8%) [p<0.0001], an 87% reduction. There were 4 deaths in subjects treated with placebo and no deaths in subjects treated with bamlanivimab 700 mg and etesevimab 1,400 mg together (p=0.01).
Secondary endpoints include mean change in viral load from baseline to Day 3, 5, and 7 (Figure 1).
Figure 1: SARS-CoV-2 Viral Load Change from Baseline (Mean ± SE) by Visit from the Phase 3 Portion of BLAZE-1 (700 mg bamlanivimab and 1,400 mg etesevimab).
The median time to sustained symptom resolution as recorded in a trial specific daily symptom diary was 8 days for subjects treated with bamlanivimab 700 mg and etesevimab 1,400 mg together as compared with 10 days for subjects treated with placebo (p=0.009). Symptoms assessed were cough, shortness of breath, feeling feverish, fatigue, body aches and pains, sore throat, chills, and headache. Sustained symptom resolution was defined as absence of any of these symptoms, except for allowance of mild fatigue and cough, in two consecutive assessments.
Phase 3 Data from BLAZE-1 (bamlanivimab 2,800 mg and etesevimab 2,800 mg)
Subjects were treated with a single infusion of bamlanivimab 2,800 mg and etesevimab 2,800 mg (N=518) or placebo (N=517). All of the patients enrolled in these dose arms met the criteria for high-risk adults (≥18 years of age) that included at least one of the following: age ≥65 years of age, BMI ≥35, chronic kidney disease, diabetes, immunosuppressive disease, immunosuppressant treatment, or age ≥55 years with cardiovascular disease, hypertension, chronic pulmonary disease or other chronic respiratory disease. Participants ages 12-17 years were also enrolled in the trial (4 [0.8%] were treated with bamlanivimab and etesevimab and 7 [1.4%] were treated with placebo), and met high-risk criteria as defined in the trial protocol.
Bamlanivimab 2,800 mg and etesevimab 2,800 mg is not an authorized dosage under this EUA. The baseline demographics and disease characteristics were well balanced across treatment groups.
The primary endpoint was the proportion of subjects with COVID-19 related hospitalization (defined as ≥24 hours of acute care) or death by any cause by Day 29. Events occurred in 36 subjects treated with placebo (7%) as compared to 11 events in subjects treated with bamlanivimab 2,800 mg and etesevimab 2,800 mg together (2%) [p<0.001], a 70% reduction. There were 10 deaths in subjects treated with placebo and no deaths in subjects treated with bamlanivimab 2,800 mg and etesevimab 2,800 mg together (p<0.001).
Pediatric Patients <18 Years
The safety and efficacy of bamlanivimab and etesevimab together was evaluated in a total of 125 pediatric subjects enrolled in the Phase 2/3 BLAZE-1 trial (NCT04427501), in which subjects were treated for mild to moderate COVID-19. Pediatric subjects were not hospitalized, and treatment was initiated within 3 days of obtaining the clinical sample for the first positive SARS-CoV-2 viral infection determination. All pediatric subjects met the criteria for high-risk (as defined in Section 2). Pediatric patients weighing 40 kg or more received the same dose as adults (700 mg bamlanivimab and 1,400 mg etesevimab). Pediatric subjects weighing less than 40 kg received weight-based dosing.
Of the 125 pediatric subjects, 33 subjects ages 12 to <18 were evaluated in double-blind, placebo-controlled Phase 3 cohorts of BLAZE-1, and 1 subject age 12 to <18 was evaluated in a controlled addendum to BLAZE-1. Of the 33 pediatric subjects, 14 received placebo, 14 received the authorized dose or a higher dose for their age, and 5 received a lower dose than authorized for their age. A total of 91 pediatric subjects were evaluated in an open-label addendum to BLAZE-1, with 40 subjects ages 12 to <18, 36 ages 6 to <12, 10 ages 2 to <6, and 5 ages 0 to <2. The youngest participant in the trial was 10 months of age and weighed 8.6 kg.
At baseline, median age was 12 years; 46% of subjects were female, 38% were White, 20% were Hispanic or Latino, and 57% were Black or African American. Subjects had mild (88%) to moderate (12%) COVID-19; the mean duration of symptoms was 4 days; mean viral load by cycle threshold (CT) was 5.92 at baseline.
No pediatric subjects died or required hospitalization due to COVID-19. The change in viral load to Day 7 by dose was: -4.23 for subjects treated with 700 mg bamlanivimab and 1,400 mg etesevimab (n=9) and -4.23 for subjects receiving weight-based dosing with bamlanivimab and etesevimab (n=75).
The median time to complete symptom resolution as recorded in a trial specific daily symptom diary was 7 days for subjects treated with bamlanivimab 700 mg and etesevimab 1,400 mg (n=10) and 5 days for subjects treated with weight-based dosing of bamlanivimab and etesevimab (n=91). Symptoms assessed were shortness of breath, nasal congestion, fever, chills, sore throat, stomachache, nausea, vomiting, diarrhea, cough, tiredness, muscle or body aches, headache, new loss of smell, new loss of taste, and poor appetite or poor feeding. Complete symptom resolution was defined as absence of all symptoms at a single timepoint.
18.2 Post-Exposure Prophylaxis of COVID-19 (BLAZE-2)
The data supporting this EUA for post-exposure prophylaxis of COVID-19 are based on the final analysis of Part 1 of the Phase 3 trial BLAZE-2 (NCT04497987). The database lock occurred after all enrolled subjects completed Day 57. BLAZE-2 Part 1 is a randomized, double-blind, placebo-controlled study evaluating bamlanivimab alone for prevention of COVID-19 in residents and staff of skilled nursing facilities following a confirmed reported case of SARS-CoV-2 infection at the facility. No pediatric participants were enrolled.
All participants in Part 1 were randomized and treated with a single infusion of bamlanivimab 4,200 mg or placebo. Results of baseline testing for SARS-CoV-2 were not known until after the therapy was administered. Those with a positive baseline SARS-CoV-2 RT-PCR test were included in the Treatment Population (N=132) and those with a negative test were included in the Prevention Population (N=966). Individuals in these populations were also required to have a baseline negative SARS-CoV-2 serology test; those who tested positive were only included in the overall safety population.
Data are presented for the Prevention Population only. No data were collected on the type or extent of exposure to the index case in the Prevention Population.
In the overall Prevention Population (N=484 for bamlanivimab 4,200 mg and N=482 for placebo) at baseline, the median age was 53 years (with 29% of subjects aged 65 or older); 75% of subjects were female, 89% were White, 5% were Hispanic or Latino, and 8% were Black. The baseline demographics and disease characteristics were well balanced across bamlanivimab and placebo treatment groups.
The primary endpoint (cases of symptomatic COVID-19 by Day 57) was assessed after all participants in the Prevention Population reached 8 weeks of follow-up, and analysis were adjusted for facility, sex, and role within facility (resident/staff). There were 114 cases of symptomatic COVID-19, with a lower frequency occurring in participants treated with bamlanivimab as compared to placebo (residents and staff; adjusted odds ratio 0.43; p<0.001) reducing the risk of being infected with COVID-19 by up to 57%. As a supplementary analysis, the time to symptomatic COVID-19 is shown for each arm in Figure 2. Four COVID-19-related deaths were reported in the overall Prevention Population; all occurred in the placebo arm (0.8%). No COVID-19-related deaths occurred in the bamlanivimab arm.
Figure 2: Time to symptomatic COVID-19 in the overall prevention population (residents and staff).
For the pre-specified subgroup of nursing home residents, there were 45 cases of symptomatic COVID-19, with a lower frequency in those treated with bamlanivimab versus placebo (adjusted odds ratio 0.20; p<0.001), reducing the risk of being infected with COVID-19 by up to 80%. The time to symptomatic COVID-19 in nursing home residents is shown by treatment arm in Figure 3. In this same cohort of residents within the Prevention Population, 6 deaths due to any cause occurred in residents treated with placebo (4.3%) and 5 deaths due to any cause occurred in residents treated with bamlanivimab (3.1%).
Figure 3: Time to symptomatic COVID-19 in residents only.
For the post-hoc subgroup of patients who met the high risk criteria (all residents and all high risk staff7), there were 75 cases of symptomatic COVID-19, with a lower frequency in those treated with bamlanivimab versus placebo (adjusted odds ratio 0.28; nominal p<0.001), reducing the risk of being infected with COVID-19 by up to 72%.
For the post-hoc subgroup of staff who did not meet high risk criteria, there were 39 cases of symptomatic COVID-19, with less evidence of a preventative effect for bamlanivimab versus placebo (adjusted odds ratio 0.64; nominal p=0.26).
- 7
- All high risk participants in the Prevention Population were either residents in a skilled nursing or assisted living facility, or staff in a skilled nursing or assisted living facility who satisfied at least 1 of the following at the time of screening: were ≥65 years of age, had a BMI ≥35, had CKD, had diabetes, had immunosuppressive disease, were currently receiving immunosuppressive treatment, OR were ≥55 years of age AND had cardiovascular disease, OR hypertension, OR COPD or other chronic respiratory disease.
19. How is Bamlanivimab and Etesevimab Injection supplied
How Supplied
UNDER THIS EUA, BAMLANIVIMAB AND ETESEVIMAB MUST BE ADMINISTERED TOGETHER.
Bamlanivimab
Bamlanivimab injection is a sterile, preservative-free clear to opalescent and colorless to slightly yellow to slightly brown solution supplied in a vial.
Etesevimab
Etesevimab injection is a sterile, preservative-free clear to opalescent and colorless to slightly yellow to slightly brown solution supplied in a vial.
Bamlanivimab and etesevimab are supplied as:
| Antibody | Concentration | Package Size | NDC |
| Bamlanivimab | 700 mg/20 mL (35 mg/mL) | one vial per carton |
0002-7910-01 |
| Etesevimab | 700 mg/20 mL (35 mg/mL) | one vial per carton |
0002-7950-01 |
Storage and Handling
Bamlanivimab is preservative-free. Discard unused portion.
Etesevimab is preservative-free. Discard unused portion.
Store unopened vials in a refrigerator at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light.
FDA has authorized an extension to the shelf-life (i.e., expiration date) of both bamlanivimab and etesevimab following a thorough review of data submitted by Eli Lilly and Company. The extension applies to all unopened vials of bamlanivimab and etesevimab that have been held in accordance with storage conditions. Confirm the shelf-life of unopened vials of bamlanivimab and etesevimab by batch number at the FDA EUA website under the Drug and Biological Therapeutic Products bamlanivimab and etesevimab. This site includes a complete listing of extended expiration dates by batch number. If the batch number on the vial/carton is not included in this listing, the product is labeled with the correct expiration date.
DO NOT FREEZE, SHAKE, OR EXPOSE TO DIRECT LIGHT.
The prepared infusion solution is intended to be used immediately. If immediate administration is not possible, store infusion solution in the refrigerator at 2°C to 8°C (36°F to 46°F) for up to 24 hours and at room temperature (20°C to 25°C [68°F to 77°F]) and for up to 7 hours, including infusion time. If refrigerated, allow the infusion solution to equilibrate to room temperature prior to administration.
20. Patient Counseling Information
Patients treated with bamlanivimab and etesevimab should continue to self-isolate and use infection control measures (e.g., wear mask, isolate, social distance, avoid sharing personal items, clean and disinfect “high touch” surfaces, and frequent handwashing) according to CDC guidelines. Also see Fact Sheet for Patients, Parents and Caregivers.
21 CONTACT INFORMATION
For additional information visit:
www.LillyAntibody.com
If you have questions, please contact:
1-855-LillyC19 (1-855-545-5921)
Literature revised January 24, 2022
Eli Lilly and Company, Indianapolis, IN 46285, USA
Copyright © 2021, 2022, Eli Lilly and Company. All rights reserved.
ETE-0009-EUA HCP-20220124
Fact Sheet for Patients, Parents and Caregivers
Emergency Use Authorization (EUA) of Bamlanivimab and Etesevimab for Coronavirus Disease 2019 (COVID-19)
You or your child are being given two medicines together called bamlanivimab and etesevimab for the treatment or post-exposure prophylaxis for prevention of coronavirus disease 2019 (COVID-19). SARS-CoV-2 is the virus that causes COVID-19. This Fact Sheet contains information to help you understand the potential risks and potential benefits of taking bamlanivimab and etesevimab.
Receiving bamlanivimab and etesevimab may help to treat COVID-19 in certain people, or help to prevent COVID-19 in certain people who have been exposed to someone infected with SARS-CoV-2 or who are at high risk of an exposure because of being in the same setting, such as nursing homes or prisons.
Read this Fact Sheet for information about bamlanivimab and etesevimab. Talk to your or your child’s healthcare provider if you have questions. It is your choice if you or your child receive bamlanivimab and etesevimab or you may stop them at any time.
What is COVID-19?
COVID-19 is caused by a virus called a coronavirus, SARS-CoV-2. People can get COVID-19 through contact with another person who has the virus.
COVID-19 illnesses have ranged from very mild (including some with no reported symptoms) to severe, including illness resulting in death. While information so far suggests that most COVID-19 illness is mild, serious illness can happen and may cause some of your or your child’s other medical conditions to become worse. People of all ages with severe, long-lasting (chronic) medical conditions like heart disease, lung disease, and diabetes, for example, and other conditions including obesity, seem to be at higher risk of being hospitalized for COVID-19. Older age, with or without other conditions, also places people at higher risk of being hospitalized for COVID-19.
What are the symptoms of COVID-19?
The symptoms of COVID-19 include fever, cough, and shortness of breath, which may appear 2 to 14 days after exposure. Serious illness including breathing problems can occur and may cause other medical conditions to become worse.
What are bamlanivimab and etesevimab?
Bamlanivimab and etesevimab are investigational medicines used together in adults and children who are at high risk for developing severe COVID-19, including hospitalization or death for:
- treatment of mild to moderate symptoms of COVID-19, OR
-
post-exposure prophylaxis for prevention of COVID-19 in persons who are:
- not fully vaccinated against COVID-19 (Individuals are considered to be fully vaccinated 2 weeks after their second dose in a 2-dose series [such as the Pfizer or Moderna vaccines], or 2 weeks after a single-dose dose vaccine [such as Johnson & Johnson’s Janssen vaccine]), or
- are not expected to build up enough of an immune response to the complete COVID-19 vaccination (for example, someone with immunocompromising conditions, including someone who is taking immunosuppressive medications), and
- have been exposed to someone who is infected with SARS-CoV-2. Close contact with someone who is infected with SARS-CoV-2 is defined as being within 6 feet for a total of 15 minutes or more, providing care at home to someone who is sick, having direct physical contact with the person (hugging or kissing, for example), sharing eating or drinking utensils, or being exposed to respiratory droplets from an infected person (sneezing or coughing, for example). For additional details, go to https://www.cdc.gov/coronavirus/2019-ncov/if-you-are-sick/quarantine.html, or
- someone who is at high risk of being exposed to someone who is infected with SARS-CoV-2 because of occurrence of SARS-CoV-2 infection in other individuals in the same institutional setting (for example, nursing homes, prisons).
Bamlanivimab and etesevimab are investigational because they are still being studied. There is limited information known about the safety or effectiveness of using bamlanivimab and etesevimab to treatment or prevention of COVID-19. Bamlanivimab and etesevimab are not authorized for pre-exposure prophylaxis for prevention of COVID-19.
The FDA has authorized the emergency use of bamlanivimab and etesevimab together for the treatment of COVID-19 and the post-exposure prophylaxis for prevention of COVID-19 under an Emergency Use Authorization (EUA). For more information on EUA, see the section “What is an Emergency Use Authorization (EUA)?” at the end of this Fact Sheet.
What should I tell the healthcare provider before I or my child receive bamlanivimab and etesevimab?
Tell the healthcare provider about all of your or your child’s medical conditions, including:
- Having any allergies
- Having received a COVID-19 vaccine
- Having any serious illnesses
- Are pregnant or plan to become pregnant
- Are breastfeeding or plan to breastfeed
- Are taking any medications (prescription, over-the-counter, vitamins, and herbal products)
How are bamlanivimab and etesevimab given?
- Bamlanivimab and etesevimab are given at the same time through a vein (intravenous or IV).
- One dose of bamlanivimab and etesevimab will be given by IV infusion. The infusion will take 16 – 60 minutes or longer. Your or your child’s healthcare provider will determine the duration of the infusion.
What are the important possible side effects of bamlanivimab and etesevimab?
Possible side effects of bamlanivimab and etesevimab are:
- Allergic reactions. Allergic reactions can happen during and after infusion with bamlanivimab and etesevimab. Tell your or your child’s healthcare provider right away if any of the following signs and symptoms of allergic reactions occur: fever, chills, nausea, headache, shortness of breath, low or high blood pressure, rapid or slow heart rate, chest discomfort or pain, weakness, confusion, feeling tired, wheezing, swelling of the lips, face, or throat, rash including hives, itching, muscle aches, feeling faint, dizziness, and sweating. These reactions may be severe or life threatening.
- Worsening of COVID-19 symptoms after bamlanivimab and etesevimab therapy for active infection: You or your child may experience new or worsening symptoms after infusion for mild to moderate COVID-19, including fever, difficulty breathing, rapid or slow heart rate, tiredness, weakness or confusion. If these occur, contact your or your child’s healthcare provider or seek immediate medical attention as some of these events have required hospitalization. It is unknown if these events are related to treatment or are due to the progression of COVID-19.
The side effects of getting any medicine by vein may include brief pain, bleeding, bruising of the skin, soreness, swelling, and possible infection at the infusion site.
These are not all the possible side effects of bamlanivimab and etesevimab. Not a lot of people have been given bamlanivimab and etesevimab. Serious and unexpected side effects may happen. Bamlanivimab and etesevimab are still being studied so it is possible that all of the risks are not known at this time.
It is possible that bamlanivimab and etesevimab could interfere with your or your child’s body’s own ability to fight off a future infection of SARS-CoV-2. Similarly, bamlanivimab and etesevimab may reduce the body’s immune response to a vaccine for SARS-CoV-2. Specific studies have not been conducted to address these possible risks. Talk to your or your child’s healthcare provider if you have any questions.
What other treatment choices are there?
Like bamlanivimab and etesevimab, FDA may allow for the emergency use of other medicines to treat people with COVID-19. Go to https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization for information on the emergency use of other medicines that are not approved by FDA to treat people with COVID-19. Your or your child’s healthcare provider may talk with you about clinical trials you or your child may be eligible for.
It is your choice whether you or your child should be treated or not to be treated with bamlanivimab and etesevimab. Should you decide that you or your child should not receive bamlanivimab and etesevimab or stop it at any time, it will not change your or your child’s standard medical care.
What other prevention choices are there?
Vaccines to prevent COVID-19 are approved or available under Emergency Use Authorization. Use of bamlanivimab and etesevimab does not replace vaccination against COVID-19.
Like bamlanivimab and etesevimab, FDA may allow for the emergency use of other medicines for post-exposure prophylaxis for prevention of COVID-19. Go to https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization for information on the emergency use of other medicines that are not approved by FDA for post-exposure prophylaxis for prevention of COVID-19. The healthcare provider may talk with you about clinical trials you or your child may be eligible for.
Bamlanivimab and etesevimab are not authorized for pre-exposure prophylaxis for prevention of COVID-19.
What if I am pregnant or breastfeeding?
There is limited experience treating pregnant women or breastfeeding mothers with bamlanivimab and etesevimab. For a mother and unborn baby, the benefit of receiving bamlanivimab and etesevimab may be greater than the risk from the treatment. If pregnant or breastfeeding, discuss your options and specific situation with your healthcare provider.
How do I report side effects with bamlanivimab and etesevimab?
Tell the healthcare provider right away if you or your child have any side effect that bothers you or your child, or does not go away.
Report side effects to FDA MedWatch at www.fda.gov/medwatch, call 1-800-FDA-1088, or contact Eli Lilly and Company at 1-855-LillyC19 (1-855-545-5921).
How can I learn more?
- Ask your or your child’s healthcare provider
- Visit www.LillyAntibody.com
- Visit https://www.covid19treatmentguidelines.nih.gov/
- Contact your local or state public health department
What is an Emergency Use Authorization (EUA)?
The United States FDA has made bamlanivimab and etesevimab available under an emergency access mechanism called an EUA. The EUA is supported by a Secretary of Health and Human Service (HHS) declaration that circumstances exist to justify the emergency use of drugs and biological products during the COVID-19 pandemic.
Bamlanivimab and etesevimab have not undergone the same type of review as an FDA-approved product. In issuing an EUA under the COVID-19 public health emergency, the FDA must determine, among other things, that based on the totality of scientific evidence available, it is reasonable to believe that the product may be effective for diagnosing, treating, or preventing COVID-19, or a serious or life-threatening disease or condition caused by COVID-19; that the known and potential benefits of the product, when used to diagnose, treat, or prevent such disease or condition, outweigh the known and potential risks of such product; and that there are no adequate, approved and available alternatives. All of these criteria must be met to allow for the medicine to be used in the treatment of COVID-19 or prevention of COVID-19 during the COVID-19 pandemic.
The EUA for bamlanivimab and etesevimab together is in effect for the duration of the COVID-19 declaration justifying emergency use of these products, unless terminated or revoked (after which the products may no longer be used).
Literature revised December 3, 2021
Eli Lilly and Company, Indianapolis, IN 46285, USA
ETE-0005-EUA PAT-20211203
PACKAGE LABEL- etesevimab Injection 700 mg/20 mL (35 mg/mL) Vial Carton
NDC 0002-7950-01
Lilly
etesevimab injection
700 mg/20 mL
(35 mg/mL)
For Intravenous Infusion Only
Must dilute before use
Single-Dose Vial: Discard Unused Portion
For use under Emergency Use Authorization (EUA).
MUST ADMINISTER WITH BAMLANIVIMAB
Copyright © 2021, Eli Lilly and Company. All rights reserved.
В настоящее время имеются предпосылки, такие как данные многоцентровых клинических исследований фаз 2 и 3, чтобы рекомендовать препараты сотровимаб и бамланивимаб+этесевимаб на догоспитальном этапе для амбулаторных пациентов с положительным тестом на COVID-19 и в первые 10 дней от появления первых симптомов. Необходимы дальнейшие исследования и долгосрочные данные текущих исследований, чтобы подтвердить или опровергнуть первоначальные результаты и понять, как появление новых вариантов вирусов может влиять на эффективность моноклональных антител, нейтрализующих SARS-CoV-2.
Введение
Моноклональные антитела (mAbs, МАт) – это молекулы, полученные лабораторным путем из В-клеток пациента, переболевшего инфекцией SARS-CoV-2. Они исследуются как потенциальная терапия новой коронавирусной инфекции (COVID-19) [1]. Вируснейтрализующим действием в отношении SARS-CoV-2 обладают искусственные МАт. МАт человека класса IgG1, связываясь с неперекрывающимися эпитопами рецептор-связывающего домена S-белка, блокируют взаимодействие S-белка SARS-CoV-2 с ангиотензинпревращающим ферментом-2 (АПФ2), что приводит к подавлению инфицирования клеток хозяина и останавливает репликацию вируса [2]. На примере сотровимаба также известно, что МАт связываются с участком вируса, содержащим гликан N343, который высококонсервативен в подроде Sarbecovirus (подрод коронавирусов). Эта область не конкурирует со связыванием АПФ и не перекрывается мутациями, которые сейчас наблюдаются у SARS-Cov-2 [3].
Характеристика препаратов
Поскольку МАт являются относительно новым классом препаратов для лечения инфекции, вызванной SARS-Cov-2, в обзор были включены данные о трех препаратах, ввоз которых в настоящее время разрешен на территорию РФ (бамланивимаб, этесевимаб, сотровимаб). С учетом того, что в августе 2021 г. Кокрановским сообществом был опубликован систематический обзор по данным препаратам [1], сначала будет представлена характеристика каждого из них наряду с данными, которые не вошли в обзор, а далее сам обзор с выводами. В связи с тем, что с мая 2021 г. препарат бамланивимаб назначается только в комбинации с этесевимабом, эти препараты будут рассмотрены совместно.
Сотровимаб. Препарат произведен британской GSK и американской VirBiotechnology [4]. В исследованиях препарат известен как Sotrovimab, а также под номерами VIR-7831, GSK4182136. На сайте https://clinicaltrials.gov (доступ 28.10.2021) найдено 9 клинических исследований (КИ) с упоминанием данного препарата [5], из них завершено только одно ключевое исследование COMET-ICE [NCT04545060] – рандомизированное многоцентровое двойное слепое плацебо-контролируемое для оценки безопасности и эффективности МАт VIR-7831 (сотровимаб) для раннего лечения COVID-19 у негоспитализированных пациентов [6]. Первые промежуточные результаты опубликованы 27 октября 2021 г., поэтому часть данных не попала в Кокрановский обзор. В этом двойном слепом исследовании третьей фазы исследователи случайным образом распределили в соотношении 1:1 в группу лечения и в группу контроля (плацебо) негоспитализированных пациентов с симптомами COVID-19 (≤5 дней после появления симптомов) и по крайней мере одним фактором риска при прогрессировании заболевания. Критерием включения служили пациенты от 18 до 55 лет с сопутствовавшими заболеваниями (факторами риска) и пациенты старше 55 лет независимо от факторов риска. Факторы риска: диабет, ожирение (индекс массы тела [ИМТ]>35), хроническая болезнь почек, застойная сердечная недостаточность (2-го класса и выше), хроническая обструктивная болезнь легких. Также у пациентов должно быть наличие подтвержденного COVID-19, сатурация 94% и более и по одному, или более из следующих симптомов: лихорадка, озноб, кашель, боль в горле, недомогание, головная боль, боль в суставах или мышцах, изменение запаха или вкуса, рвота, диарея, одышка при физической нагрузке. Исключались госпитализированные пациенты или те, которым планируется госпитализация в ближайшие 24 часа, а также с симптомами, соответствующими тяжелой форме COVID-19, которые определяются одышкой в состоянии покоя, респираторным дистресс-синдромом или потребностью в кислороде. Пациентам, включенным в исследование, проводилась однократная инфузия сотровимаба в дозе 500 мг или плацебо. Первичным результатом эффективности была госпитализация (более 24 часов) по поводу любой причины или смерти в течение 29 дней после рандомизации. В этом промежуточном анализе с участием 583 пациентов (291 в группе сотровимаба и 292 в группе плацебо); 3 (1%) пациента в группе сотровимаба по сравнению с 21 (7%) пациентом в группе плацебо имели прогрессирование заболевания, приведшее к госпитализации или смерти (снижение отношения рисков [ОР]=85%, 97,24% доверительный интервал [ДИ]: 44–96; р=0,002). В группе плацебо пять пациентов были госпитализированы в отделение интенсивной терапии, в т.ч. один умер на 29-й день. Безопасность была оценена у 868 пациентов (430 в группе сотровимаба и 438 в группе плацебо). О побочных эффектах сообщили 17% пациентов в группе сотровимаба и 19% пациентов в группе плацебо; серьезные побочные эффекты при приеме сотровимаба встречались реже, чем при приеме плацебо (у 2 и 6% пациентов соответственно). Исследователи сделали вывод, согласно которому сотровимаб снижает риск прогрессирования заболевания среди пациентов с высоким риском COVID-19 от легкой до умеренной степени тяжести. Сигналов безопасности выявлено не было [6]. Также обращают на себя внимание другие исследования сотровимаба: COMET-PEAK [NCT04779879], ACTIV-3-TICO [NCT04501978] и BLAZE-4 [NCT0463440 [9]. Они еще не завершены, официальные публикации отсутствуют, а часть данных также попала в Кокрановсий обзор, выдержки из которого представлены ниже.
Комбинация бамланивимаб+этесевимаб. Бамланивимаб и этесевимаб являются рекомбинантными нейтрализующими МАт человека IgG1κ к S-белку SARS-CoV-2, которые блокируют связывание данного белка с рецептором АПФ2 человека с целью предотвращения дальнейшего проникновения вируса в клетку. Бамланивимаб и этесевимаб связываются с разными участками рецептор-связывающего домена (RBD) S-белка [7].
В РКИ показано достоверное снижение вирусной нагрузки на 11-й день лечения при применении комбинации бамланивимаб+этесевимаб по сравнению с монотерапией бамланивимабом [8]. Частота госпитализаций пациентов или обращений за неотложной помощью в связи с COVID-19 составила 9 (5,8%) случаев при приеме плацебо, 1 (1%), 2 (1,9%) случая, 2 (2%) случая при приеме банланивимаба в дозе 700 мг, 2800 и 7000 мг соответственно и 1 (0,9%) случай при приеме комбинации бамланивимаб+этесевимаб. Реакции гиперчувствительности немедленного типа развились у 9 пациентов (6 на бамланивимабе, 2 – на комбинированной терапии и 1 при приеме плацебо). Летальных исходов не зарегистрировано.
На сайте www.clinicaltrials.gov зарегистрировано 1 сравнительное исследование (III фаза) комбинации бамланивимаб+этесевимаб с монотерапией бамланивимабом, сотровимабом и комбинацией касиривимаб+имдевимаб [NCT04790786].
Согласно препринту мета-анализа 3 РКИ и 7 когортных исследований с участием 14 461 взрослого пациента, бамланивимаб может предотвращать госпитализацию или обращение за неотложной помощью (ОР=0,41, 95% ДИ: 0,29–0,58), сокращать число госпитализаций в отделение интенсивной терапии (ОР=0,47; 95% ДИ: 0,23–0,92) и смертность от болезни (ОР=0,32; 95% ДИ: 0,13–0,77]). Комбинации двух или более МАт усиливают эффект [9].
В исследовании ACTIVE-2 при использовании бамланивимаба в монотерапии в дозировке 700 мг (в настоящее время препарат не применяется в монотерапии) могут выявляться резистентные штаммы, что может в дальнейшем быть связано с сохранением вирусной нагрузки в дыхательных путях и клиническим ухудшением. При разработке и клиническом внедрении противовирусных средств в лечение COVID-19 первоочередное внимание следует уделять тщательной вирусологической оценке [10]. По мнению некоторых авторов, широкое использование антител к SARS-CoV-2 может приводить к селекции устойчивых штаммов и потребовать усиленного геномного надзора для выявления и снижения распространения этих вариантов [11].
Кокрановский обзор исследований МАт
Поскольку основные исследования по данной проблеме представлены и описаны в Кокрановском обзоре [1], мы решили частично представить его результаты. Авторы обзора от 17.06.2021 провели поиск в MEDLINE, Embase, Кокрановском регистре исследований COVID-19, а также в трех других базах данных, кроме того, запрашивали необходимые данные у авторов публикаций.
В обзор включали только рандомизированные контролируемые исследования (РКИ), в которых оценивали МАт, нейтрализующие SARS-CoV-2, отдельно или в комбинации по сравнению с другим активным лечением, плацебо или отсутствием лечения пациентов с COVID-19. Не включались исследования по профилактическому применению МАт, нейтрализующих SARS-CoV-2. Приоритетными исходами были смертность от всех причин к 30-му и 60-му дням, клиническое прогрессирование, качество жизни, госпитализация, нежелательные явления (НЯ) и серьезные нежелательные явления (СНЯ). Достоверность доказательств оценили с помощью системы GRADE.
В обзор попали шесть РКИ с запланированными сроками завершения в период с июля 2021 по декабрь 2021 г., которые предоставили результаты от 17 495 участников (мы приводим данные по препаратам бамланивимаб, сотровимаб, этесевимаб). Целевые размеры выборки варьировались от 1020 до 10 тыс. участников. Средний возраст составлял от 42 до 53 лет в четырех РКИ с участием негоспитализированных пациентов и 61 год в двух РКИ с участием госпитализированных пациентов. В нескольких исследованиях оценивались монопрепараты бамланивимаб (N=465), сотровимаб (N=868), а также комбинация бамланивимаб+этесевимаб (N=1035).
Сравнение бамланивимаба с плацебо. К 29-му дню в исследовании не было летальных исходов. К 29-му дню наблюдения были госпитализированы 9 человек из 156 в группе плацебо по сравнению с 1 из 101 в группе пациентов, получавших бамланивимаб в дозе 700 мг (ОР=0,17, 95% ДИ: 0,02–1,33]), 2 из 107 в группе пациентов, получавших 2,8 г (ОР=0,32, 95% ДИ: 0,07–1,47]) и 2 из 101 в группе пациентов, получавших бамланивимаб в дозе 7000 мг (ОР=0,34, 95% ДИ: 0,08–1,56). При применении 700 мг, 2800, 7000 мг бамланивимаба частота НЯ может быть аналогичной плацебо (ОР=0,99, 95% ДИ: 0,66–1,50; ОР=0,90, 95% ДИ: 0,59–1,38; ОР=0,81, 95% ДИ: 0,52–1,27). Влияние на риск СНЯ не определено. О клиническом прогрессировании/улучшении симптомов или развитии тяжелых симптомов не сообщалось.
Сравнение комбинации бамланивимаб+этесевимаб с плацебо. К 30-му дню было зарегистрировано 10 смертей в группе плацебо и ни одного случая смерти в группе бамланивимаб+этесевимаб (ОР=0,05, 95% ДИ: 0,00–0,81). Бамланивимаб+этесевимаб может снижать число госпитализаций к 29-му дню (ОР=0,30, 95% ДИ: 0,16–0,59), приводить к небольшому увеличению числа НЯ любой степени (ОР=1,15, 95% ДИ: 0,83–1,59) и увеличивать СНЯ (ОР=1,40, 95% ДИ: 0,45–4,37).
О клиническом прогрессировании/улучшении симптомов или развитии тяжелых симптомов не сообщалось.
Сотровимаб по сравнению с плацебо. Не установлено, влияет ли сотровимаб на смертность (ОР=0,33, 95% ДИ: 0,01–8,18), а также потребность в инвазивной механической вентиляции или смерть (ОР=0,14, 95% ДИ: 0,01–2,76). Сотровимаб может снижать потребность в кислороде (ОР=0,11, 95% ДИ: 0,02–0,45), риск госпитализации или смерти к 30-му дню (ОР=0,14, 95% ДИ: от 0,04 до 0,48), НЯ 3–4-й степеней (ОР=0,26, 95% ДИ: 0,12–0,60), СНЯ (ОР=0,27, 95% ДИ: 0,12–0,63) и может иметь незначительное влияние или его не оказывать на риск НЯ любой степени (ОР= 0,87, 95% ДИ: 0,66–1,16).
В заключение Кокрановского обзора сделан вывод [1], согласно которому уровень доказательств для всех негоспитализированных лиц низкий, а для госпитализированных – от очень низкого до умеренного. Авторы считают, что имеющихся данных недостаточно для того, чтобы сделать значимые выводы относительно лечения с помощью МАт, нейтрализующих SARS-CoV-2. Необходимы дальнейшие исследования и долгосрочные данные текущих исследований, чтобы подтвердить или опровергнуть эти первоначальные результаты и понять, как появление новых вариантов вирусов может влиять на эффективность МАт, нейтрализующих SARS-CoV-2.
Возможность применения моноклональных антител в отношении пациентов, получивших вакцины против SARS-COV-2
Данные о применении МАт пациентами, привитыми против SARS-CoV-2, ограничены. Изучалась эффективность МАт у 1395 пациентов, заболевших COVID-19 после полного курса вакцинации [12], из них 37,8% (n=527) получали МАт против S-белка и 62,2% (n=868) не получали этого лечения. Пациенты принимали МАт (казиривимаб–имдевимаб или бамланивимаб+этесевимаб) в среднем через 5 дней с момента появления симптомов и через 2 дня с момента тестирования большинство пациентов принимали казиривимаб+имдевимаб, что ограничивает возможность обобщения результатов на другие МАт. В целом частота госпитализации составила 2,65% среди пациентов, получавших МАт, по сравнению с 10,7% среди тех, кто не получал терапии (ОШ=0,227, 95% ДИ: 0,128–0,403; p<0,001).
С поправкой на критерии MASS включения в терапию МАт она была связана с более низким уровнем госпитализации (ОШ=0,14, 95% ДИ: 0,079–0,265]; p<0,0001). NNT для предотвращения одной госпитализации коррелировало со степенью сопутствующей патологии и составило 255 среди вакцинированных лиц без сопутствующих заболеваний высокого риска и от 3 до 8 среди пациентов с множественными заболеваниями высокого риска. У 61 (4,4%) пациента развилась гипоксия, и им потребовалась кислородотерапия. Лечение МАт против S-белка было связано с более низкими показателями гипоксии (0,95 против 6,45%; ОШ=0,139, 95% ДИ: 0,055–0,349; p<0,001). Большинство (n=56, 91,8%) пациентов, которым требовалась кислородотерапия, не получали Мат, 5 (0,36%) пациентам потребовалась госпитализация в отделение интенсивной терапии, из них ни один не получал МАт. Из 1395 вакцинированных пациентов с COVID-19 не было летальных случаев.
Особенности применения МАт при COVID-19, показания и противопоказания, дозы
В настоящий момент многие страны разрешили экстренное применение препаратов МАт сотровимаб и комбинации бамланивимаб+этесевимаб. Препараты сотровимаб и комбинация бамланивимаб+этесевимаб в разное время были разрешены в странах Европы и США по ускоренным процедурам. Сотровимаб был одобрен Американской FDA (с 26.05.2020) [13], Европейским медицинским агентством EMA (с 21.05.2020) [14], Медицинским департаментом Австралийского управления терапевтических товаров (TGA) (с 21.08.2021) [15]. Бамланивимаб+этесевимаб был одобрен к применению регуляторными органами FDA, США (c 09.02.2021 с пометкой к использованию только в тех штатах, территориях и юрисдикциях США, в которых комбинированная частота вариантов, устойчивых к бамланивимабу и этесевимабу, вводимых вместе, меньше или равна 5%) [16], Европейским медицинским агентством EMA (c 05.03. 2021) [17].
Назначение комбинации бамланивимаб+этесевимаб и сотровимаба одобрено Временными методическими рекомендациями по профилактике, диагностике и лечению новой коронавирусной инфекции (COVID-19), 13-е издание, при наличии решения врачебной комиссии и разрешения на временное обращение данных препаратов [2].
Препараты сотровимаб и комбинация бамланивимаб+этесевимаб имеют схожие показания и целевые категории пациентов: они одобрены к применению в первые 10 дней заболевания для лечения легкой и средней степеней тяжести COVID-19 у взрослых и детей (12 лет и старше, с массой тела не менее 40 кг) с положительным результатом прямого тестирования на вирус SARS-CoV-2 и высоким риском прогрессирования до тяжелой формы Covid-19, включая госпитализацию или смерть [2, 13–15, 18, 19].
Критерии высокого риска [18, 19]:
- пожилой возраст (≥65 лет);
- ожирение или избыточная масса тела (например, взрослые с ИМТ>25 кг/м2 или если лица в возрасте 12–17 лет имеют ИМТ≥85-го процентиля для своего возраста и пола по данным CDC (https://www.cdc.gov/growthcharts/clinical_charts.htm);
- следующие заболевания или состояния: беременность, хроническое заболевание почек, диабет, иммунодепрессивное заболевание или прием иммунодепрессантов, сердечно-сосудистые заболевания (включая врожденные пороки сердца) или артериальную гипертензию, хронические заболевания легких (например, хроническая обструктивная болезнь легких, астма – от средней до тяжелой, интерстициальное заболевание легких, муковисцидоз и легочная гипертензия), серповидно-клеточная анемия, нарушения нервного развития (например, церебральный паралич) или другие отягощающие состояния (например, генетические или метаболические синдромы и тяжелые врожденные аномалии);
- наличие зависимости от медицинских приборов (например, трахеостомия, гастростомия или вентиляция с положительным давлением, не связанная с COVID-19).
Помимо этого с сентября 2021 г. комбинация бамланивимаб+этесевимаб получила ускоренное одобрение FDA для постконтактной профилактики COVID-19 у взрослых пациентов и детей с 12 лет (с массой тела от 40 кг) с высоким риском прогрессирования COVID-19 в тяжелую форму, включая госпитализацию или смерть, которые не полностью вакцинированы или от которых не ожидается адекватного иммунного ответа после полного курса вакцинации против SARS-CoV-2 (например, пациенты с иммунодефицитными состояниями, в т.ч. принимающие иммунодепрессанты) и которые:
- контактировали с человеком, инфицированным SARS-CoV-2, в соответствии с критериями тесного контакта Центра по контролю и профилактике заболеваний (CDC) либо
- имеют высокие риски контакта с человеком, инфицированным SARS-CoV-2, из-за возникновения инфекции SARS-CoV-2 у других лиц в том же учреждении (например, в домах престарелых, тюрьмах).
Препараты сотровимаб и комбинация бамланивимаб+этесевимаб не разрешены к применению в отношении пациентов [18, 19]:
- госпитализированных в связи с COVID-19, или
- нуждающихся в кислородотерапии из-за COVID-19, или
- нуждающихся в увеличении исходного потребления кислорода из-за COVID-19 (у тех, кто находится на хронической кислородной терапии из-за сопутствующей патологии, не связанной с COVID-19).
Рекомендованный режим применения комбинации бамланивимаб+этесевимаб — однократная внутривенная инфузия 700 мг бамланивимаба и 1400 мг этесевимаба в разведении с раствором натрия хлорида 0,9% 50–250 мл в течение 21–60 минут в зависимости от объема раствора как можно раньше после получения положительного ПЦР-теста на SARS-CoV-2 либо контакта с носителем инфекции [17]. Не требуется коррекция дозы детям, пожилым пациентам, беременным и кормящим женщинам, при печеночной и почечной недостаточности.
Рекомендованный режим применения сотровимаба – однократная внутривенная инфузия 500 мг в течение 30 минут, вводимая как можно скорее после положительного ПЦР-теста на SARS-CoV-2 и в течение 10 дней с момента появления симптомов [18].
Безопасность применения МАт
Согласно информации FDA [17, 18], при приеме комбинации бамланивимаб+этесевимаб и сотровимаба возможно развитие серьезных реакций гиперчувствительности, включая анафилаксию, и инфузионных реакций, включая серьезные или опасные для жизни, в т.ч. через 24 и более часов после инфузии, что требует немедленного прекращения инфузии и/или коррекции осложнения. Симптомы инфузионных реакций: лихорадка, затрудненное дыхание, гипоксемия, озноб, аритмии, боль или дискомфорт в груди, слабость, изменение психического статуса, тошнота, головная боль, головокружение, потливость бронхоспазм, гипотензия, гипертензия, ангионевротический отек, кожные реакции, вазовагальные реакции (предобморочное состояние, обморок).
В исследовании COMET-ICE по сотровимабу были зарегистрированы единичные случаи анафилаксии, частота развития реакций гиперчувствительности составила 2%, инфузионных реакций – 1%. В исследованиях BLAZE-1 и BLAZE-4 по комбинации бамланивимаб+этесевимаб частота инфузионных реакций составила 1,1%. При применении как сотровимаба, так и комбинации бамланивимаб+этесевимаб отмечались единичные случаи анафилаксии и серьезных инфузионных реакций, потребовавших отмены инфузии и фармакологической коррекции [17, 18].
Также сообщалось о клиническом ухудшении COVID-19 после применения МАт с лихорадкой, гипоксией или усилением дыхательной недостаточности, аритмиями (например, фибрилляцией предсердий, синусовой тахикардией, брадикардией), утомляемостью и психическими нарушениями; в некоторых случаях потребовалась госпитализация. Связь этих событий с применением МАт или же с прогрессированием COVID-19 не установлена [17, 18].
Оценка влияния бамланивимаба, сотровимаба и этесевимаба на риск канцерогенеза, мутагенеза и на репродуктивную функцию не проводилась [17, 18].
Заключение
В настоящее время имеются предпосылки и перспективы, чтобы рекомендовать препараты сотровимаб и бамланивимаб+этесевимаб на догоспитальном этапе для амбулаторных пациентов. Оба препарата одобрены к применению в первые 10 дней для лечения легкой и средней степеней тяжести COVID-19 у взрослых и детей (12 лет и старше с массой тела не менее 40 кг) с положительными результатами прямого тестирования на вирус SARS-CoV-2 и высоким риском прогрессирования до тяжелой формы COVID-19, включая госпитализацию или смерть.
Необходимы дальнейшие исследования и долгосрочные данные существующих исследований, чтобы подтвердить или опровергнуть первоначальные результаты исследований и понять, как появление вариантов вирусов может влиять на эффективность МАт, нейтрализующих SARS-CoV-2
Препараты нейтрализующих МАт не рекомендуется применять пациентам, госпитализированным по поводу COVID-19, нуждающимся в кислородной поддержке, механической вентиляции или пациентам на хронической кислородной терапии из-за основного заболевания, не связанного с COVID-19.
Дополнительная информация. Представленные в ответе рекомендации служат для поддержки клинических решений, принимаемых лечащим врачом, и не заменяют клинического мышления врача, самостоятельный поиск последней научной информации, сверку с действующими инструкциями по медицинскому применению лекарственных препаратов. Информация, представленная в ответе, основана на результатах научного поиска, проведенного 05.11.2021 экспертами Центра «ФармаCOVID» на базе РМАНПО Минздрава России, Москва.
Автор для связи: Виталий Михайлович Цветов, к.м.н., клинический фармаколог, Федеральный центр сердечно-сосудистой хирургии, Челябинск, Россия; SPIN-код: 3202-7659